|
|
CURSO DE BIOLOGÍAAlejandro Porto Andión |
|
|
|
|
|
|
|
Inicio Temas de Biomoléculas Aula virtual |
|
|
|
|
![]()
TEMA 8: PROTEÍNAS.
1.-INTRODUCCIÓN.
El término proteína deriva del griego "proteos" (lo primero, lo principal) y habla de su gran importancia para los seres vivos. La importancia de las proteínas es, en un primer análisis, cuantitativa: constituyen el 50% del peso seco de la célula (15% del peso total) por lo que representan la categoría de biomoléculas más abundante después del agua.
Sin embargo su gran importancia biológica reside, más que en su abundancia en la materia viva, en el elevado número de funciones biológicas que desempeñan, en su gran versatilidad funcional y sobre todo en la particular relación que las une con los ácidos nucleicos, ya que constituyen el vehículo habitual de expresión de la información genética contenida en éstos últimos.
2.-COMPOSICIÓN DE LAS PROTEÍNAS.
Desde el punto de vista de su composición elemental todas las proteínas contienen carbono, hidrógeno, oxígeno y nitrógeno, mientras que casi todas contienen además azufre (Cabe resaltar que en azúcares y lípidos el nitrógeno sólo aparece en algunos de ellos). Hay otros elementos que aparecen solamente en algunas proteínas (fósforo, cobre, zinc, hierro, etc.).
Las proteínas son biomoléculas de elevado peso molecular (macromoléculas) y presentan una estructura química compleja. Sin embargo, cuando se someten a hidrólisis ácida, se descomponen en una serie de compuestos orgánicos sencillos de bajo peso molecular: los α-aminoácidos. Este rasgo lo comparten las proteínas con otros tipos de macromoléculas: todas son polímeros complejos formados por la unión de unos pocos monómeros o sillares estructurales de bajo peso molecular. Existen 20 α-aminoácidos diferentes que forman parte de las proteínas.
En las moléculas proteicas los sucesivos restos aminoácidos se hallan unidos covalentemente entre sí formando largos polímeros no ramificados. El tipo de enlace que los une recibe el nombre de enlace peptídico. Las cadenas de aminoácidos de las proteínas no son polímeros al azar, de longitud indefinida, cada una de ellas posee una determinada composición química, un peso molecular y una secuencia ordenada de aminoácidos.
3.-CLASIFICACIÓN DE LAS PROTEÍNAS.
Las proteínas se clasifican en dos clases principales atendiendo a su composición. Las proteínas simples u holoproteínas son las que están compuestas exclusivamente por aminoácidos. Las proteínas conjugadas o heteroproteínas son las que están compuestas por aminoácidos y otra sustancia de naturaleza no proteica que recibe el nombre de grupo prostético. Las proteínas conjugadas pueden a su vez clasificarse en función de la naturaleza de su grupo prostético. Así, se habla de glucoproteínas, cuando el grupo prostético es un glúcido, lipoproteínas cuando es un lípido, metaloproteínas cuando es un ion metálico, fosfoproteínas cuando es un grupo fosfato, etc.
Otro criterio de clasificación de las proteínas es la forma tridimensional de su molécula. Las proteínas fibrosas son de forma alargada, generalmente son insolubles en agua y suelen tener una función estructural, mientras que las proteínas globulares forman arrollamientos compactos de forma globular y suelen tener funciones de naturaleza dinámica (catalíticas, de transporte, etc).
4.-TAMAÑO DE LAS MOLÉCULAS PROTEICAS.
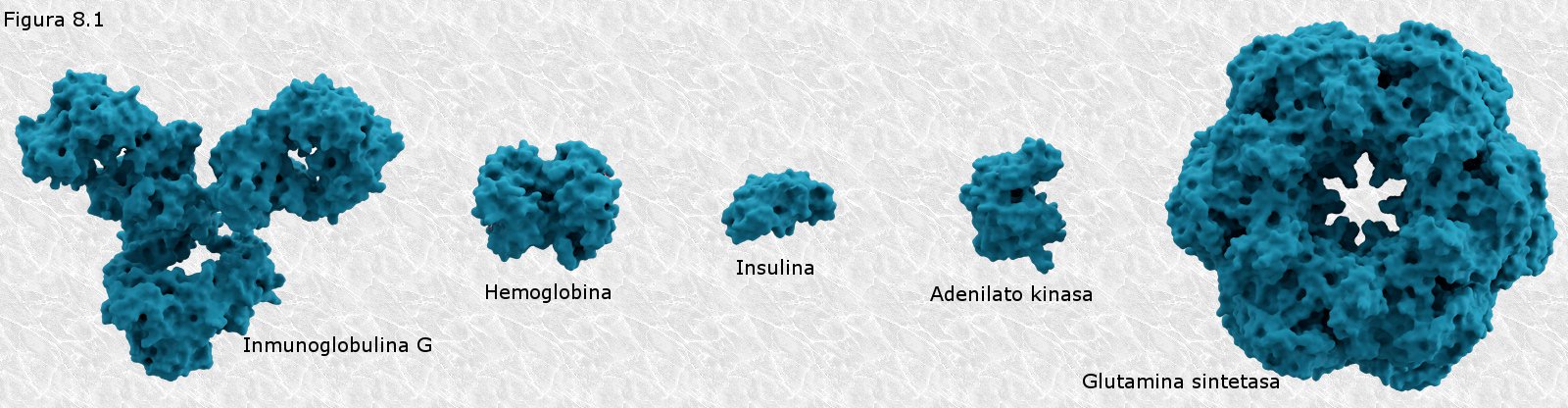
Las proteínas presentan tamaños moleculares muy variables (desde unos pocos miles de daltons a más de un millón) (ver Figura 8.1). Algunas proteínas están formadas por una sola cadena de aminoácidos, mientras que otras, llamadas proteínas oligoméricas, están formadas por varias cadenas individuales denominadas protómeros o subunidades. Se ha podido comprobar que en la mayor parte de los casos las cadenas individuales de aminoácidos presentan pesos moleculares que oscilan entre los 12.000 y los 36.000 daltons, lo que se corresponde con una longitud de entre 100 y 300 restos aminoácidos. Sin embargo existen moléculas proteicas más pequeñas como la insulina (51 aminoácidos y 5.700 daltons) y mucho más grandes como la apolipoproteína B, una proteína transportadora de colesterol, con 4.536 aminoácidos y 513.000 daltons, que representa la cadena individual de aminoácidos más grande conocida hasta la fecha. Las proteínas de mayor tamaño están formadas invariablemente por varias cadenas de aminoácidos.
5.-AMINOÁCIDOS: LOS SILLARES ESTRUCTURALES.
5.1.-CONCEPTO.
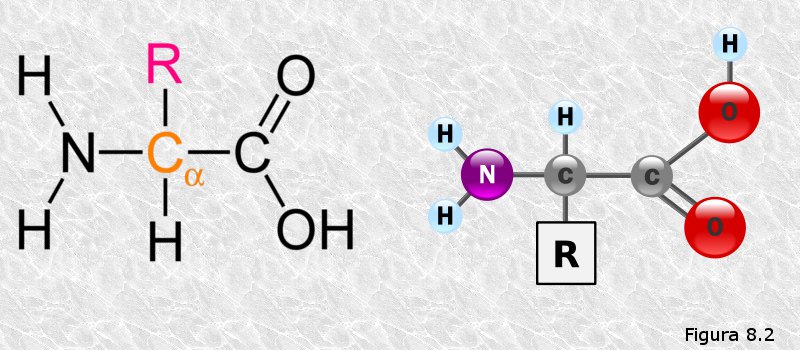
Los aminoácidos son compuestos orgánicos que poseen un grupo carboxilo y un grupo amino. Pueden ser α, β, γ, δ....aminoácidos, según el grupo amino esté unido respectivamente al primero, segundo, tercero, cuarto... átomo de carbono contando a partir del átomo de carbono del grupo carboxilo. En la naturaleza existen distintos tipos de aminoácidos que desempeñan diferentes funciones, sin embargo, los aminoácidos que forman parte de las proteínas son todos ellos α-aminoácidos.
Existen 20 α-aminoácidos diferentes que forman parte de las proteínas. Todos ellos tienen una parte de su molécula en común (formada por el átomo de carbono α unido a los grupos amino y carboxilo) y difieren entre sí en la naturaleza de la cadena lateral (habitualmente llamada grupo R). En la Figura 8.2 aparece la fórmula estructural de un α-aminoácido; en ella "R" representa la cadena lateral que difiere entre los distintos aminoácidos.
5.2.-ESTEREOISOMERÍA DE LOS AMINOÁCIDOS.
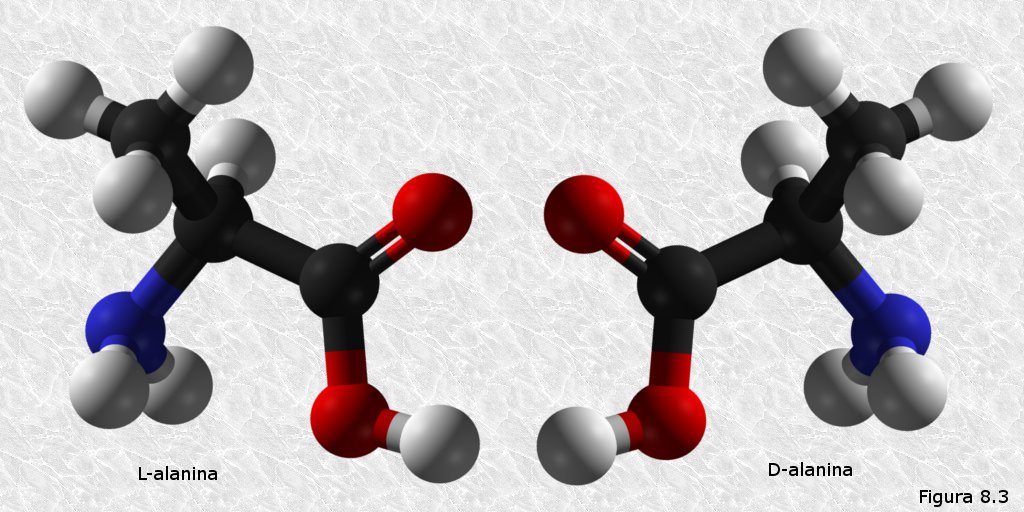
Los α-aminoácidos son compuestos quirales. En todos ellos, con la única excepción de la glicocola, el átomo de carbono α (el contiguo al grupo carboxilo) es un carbono asimétrico, es decir, un átomo de carbono unido a cuatro sustituyentes distintos. Debido a esta circunstancia, cada α-aminoácido puede existir en dos formas estereoisómeras cada una de ellas con una diferente ordenación espacial de los cuatro sustituyentes que rodean, en disposición tetraédrica, al carbono α (Figura 8.3). Estas dos formas estereoisómeras son además enantiómeros (imágenes especulares no superponibles una de la otra). La nomenclatura de las formas estereoisómeras de los α-aminoácidos se establece por convenio relacionando sus fórmulas en proyección de Fisher con la de un compuesto de referencia que es el gliceraldehido. Así, la forma D de un α-aminoácido es la que, en la fórmula en proyección de Fisher, tiene el grupo amino hacia la derecha (por analogía con el grupo hidroxilo del D-gliceraldehido), mientras que la forma L es la que lo tiene hacia la izquierda (ver Figura 8.3). Aunque existen en la naturaleza aminoácidos con configuración D que desempeñan diferentes funciones en las células, todos los aminoácidos presentes en las proteínas presentan configuración L.
Los α-aminoácidos, como todos los compuestos quirales, presentan actividad óptica, es decir, hacen girar en uno u otro sentido el plano de vibración de la luz polarizada. Así, algunos α-aminoácidos en disolución hacen girar dicho plano de vibración hacia la derecha, y se dice que son dextrógiros (+), mientras que otros lo hacen hacia la izquierda, y se dice que son levógiros (-). El carácter dextrógiro o levógiro de un α-aminoácido es independiente de la configuración D o L que presente.
5.3.-COMPORTAMIENTO ÁCIDO-BASE.
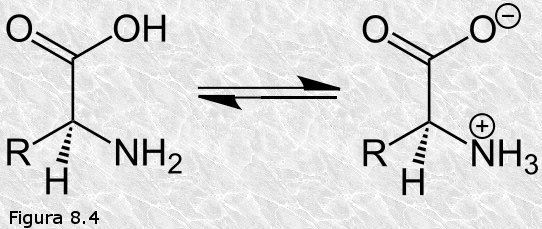 Los aminoácidos son compuestos sólidos, cristalinos, que presentan
un punto de fusión y una solubilidad en agua muy superiores a lo que
cabría esperar dado su peso molecular. Ello se debe a que los
aminoácidos existen en disolución, y cristalizan a partir de las
disoluciones, en forma de iones dipolares (Figura
8.4). A pH neutro el grupo carboxilo cede un protón y queda
cargado negativamente y el grupo amino capta un protón y queda
cargado positivamente. Así, los aminoácidos pueden comportarse como
ácidos o como bases según el pH del medio; se dice que son
sustancias anfóteras. Existe un valor de pH llamado punto
isoeléctrico (pI) para el cual el aminoácido está compensado
eléctricamente (carga neta = 0).
Los aminoácidos son compuestos sólidos, cristalinos, que presentan
un punto de fusión y una solubilidad en agua muy superiores a lo que
cabría esperar dado su peso molecular. Ello se debe a que los
aminoácidos existen en disolución, y cristalizan a partir de las
disoluciones, en forma de iones dipolares (Figura
8.4). A pH neutro el grupo carboxilo cede un protón y queda
cargado negativamente y el grupo amino capta un protón y queda
cargado positivamente. Así, los aminoácidos pueden comportarse como
ácidos o como bases según el pH del medio; se dice que son
sustancias anfóteras. Existe un valor de pH llamado punto
isoeléctrico (pI) para el cual el aminoácido está compensado
eléctricamente (carga neta = 0).
Las curvas de titulación de los aminoácidos son más complejas que las de los pares conjugados ácido-base corrientes. Esto se debe a que cada aminoácido posee dos grupos funcionales capaces de aceptar o ceder protones (amino y carboxilo), cada uno de los cuales tiene su propio pK y comportamiento ácido-base característico. Por otra parte, algunos aminoácidos presentan cadenas laterales (R) con grupos funcionales que son potenciales dadores o aceptores de protones, y que por lo tanto también influyen de manera determinante en sus propiedades ácido-base.
El comportamiento ácido-base de los aminoácidos reviste una gran importancia biológica, ya que influye a su vez en las propiedades de las proteínas de las que forman parte. Además, las técnicas para separar y analizar los aminoácidos componentes de una proteína se basan en gran medida en su comportamiento ácido-base.
5.4.-CLASIFICACIÓN DE LOS AMINOÁCIDOS.
Existen distintos criterios para clasificar los α-aminoácidos de las proteínas. Sin embargo, el más utilizado, dada su relación con la determinación de la estructura tridimensional de las mismas, es el que se basa en la naturaleza polar o no polar, con carga eléctrica o sin ella de su cadena lateral o grupo R. Así se distinguen:
a) Aminoácidos con grupo R no polar (alifáticos y aromáticos).
b) Aminoácidos con grupo R polar sin carga .
c) Aminoácidos con grupo R con carga positiva.
d) Aminoácidos con grupo R con carga negativa.
En la Tabla 8.1 se representan las fórmulas estructurales de los 20 α-aminoácidos presentes en las proteínas en las formas iónicas en las que aparecen a pH fisiológico. Todos los α-aminoácidos tienen, además de sus nombres sistemáticos, nombres simplificados apropiados para su uso común, que, en algunos casos, provienen de la fuente biológica de la cual fueron aislados inicialmente; así, la asparagina se encontró por primera vez en el espárrago, el ácido glutámico se aisló del gluten de trigo, la tirosina fue identificada en el queso (del griego tyros = queso), y la glicocola debe su nombre a su sabor dulce (del griego glycos = dulce).
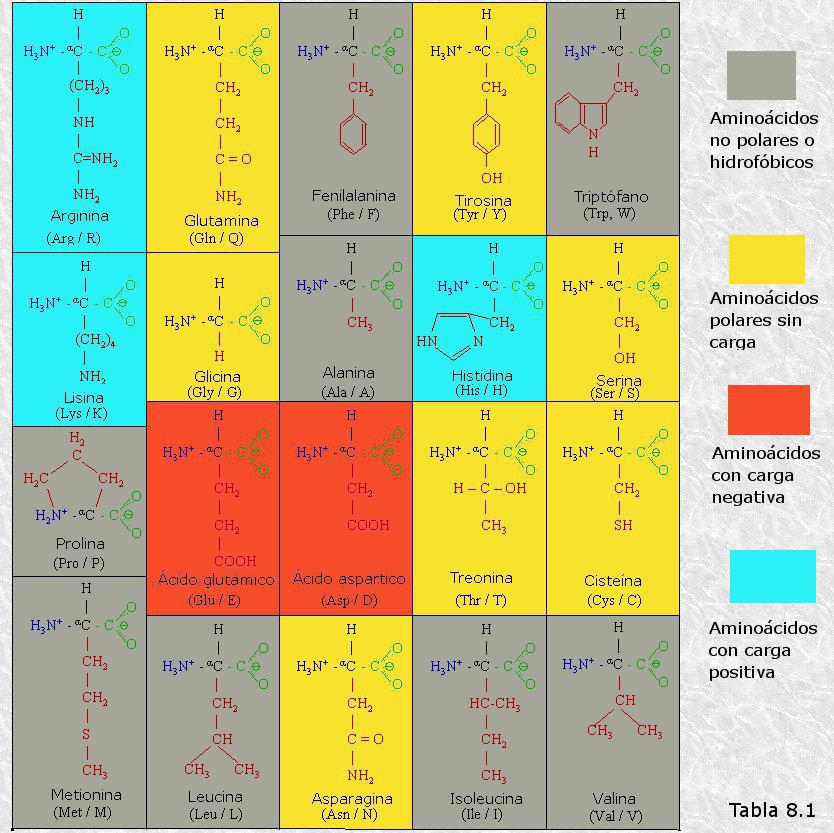
Además de los 20 α-aminoácidos que son comunes a todas las proteínas existen en algunas de ellas otros aminoácidos, denominados aminoácidos no estándar. Todos ellos derivan de alguno de los 20 aminoácidos estándar través de transformaciones químicas sencillas que se operan una vez el aminoácido de origen ha sido incorporado a la proteína. Entre ellos cabe citar la hidroxiprolina, la N-metil-lisina y la desmosina.
Por otra parte, se han encontrado en las células vivas alrededor de otros 300 aminoácidos que desempeñan diferentes funciones pero que no forman parte de las proteínas. Algunos de ellos presentan configuración D y no todos son α-aminoácidos.
6.-EL ENLACE PEPTÍDICO. LOS PÉPTIDOS.
Los aminoácidos se enlazan para formar proteínas mediante enlace peptídico. Los péptidos son compuestos formados por la unión de aminoácidos mediante enlace peptídico. El enlace peptídico es una unión covalente tipo amida sustituida que se da al reaccionar el grupo amino de un aminoácido con el grupo carboxilo de otro aminoácido con desprendimiento de una molécula de agua En la siguiente animación se puede apreciar como es el proceso de formación de un enlace peptídico.
Cuando dos aminoácidos reaccionan para formar un enlace peptídico el compuesto resultante recibe el nombre de dipéptido. Por ser el enlace peptídico una unión "cabeza-cola" (grupo amino con grupo carboxilo) un dipéptido conserva siempre un grupo amino libre, que puede reaccionar con el grupo carboxilo de otro aminoácido, y un grupo carboxilo libre, que puede reaccionar con el grupo amino de otro aminoácido (Figura 8.5). Esta circunstancia permite que mediante enlaces peptídicos se puedan enlazar un número elevado de aminoácidos formando largas cadenas que siempre tendrán en un extremo un grupo amino libre (extremo amino terminal) y en el otro un grupo carboxilo libre (extremo carboxi-terminal).
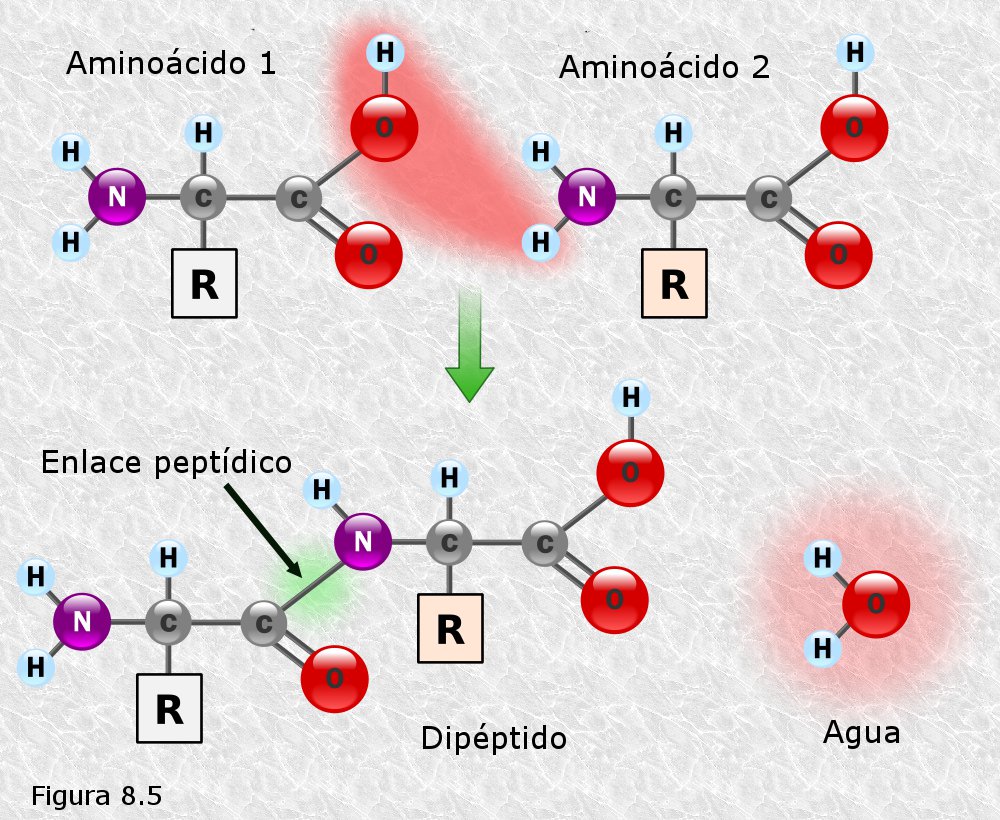
Los péptidos se clasifican según el número de restos de aminoácidos que los forman. Así los péptidos formados por 2, 3, 4,.... aminoácidos se denominan respectivamente dipéptidos, tripéptidos, tetrapéptidos... En general cuando el número de aminoácidos implicados es menor o igual a 10 decimos que se trata de un oligopéptido, cuando es mayor que 10 decimos que se trata de un polipéptido. Es también frecuente el uso del la expresión cadena polipeptídica en lugar de polipéptido. Cuando una cadena polipeptídica tiene más de 100 restos de aminoácidos (es un límite arbitrario y que no hay que tomar al pie de la letra) decimos que se trata de una proteína. Sin embargo hay que tener en cuenta que existen proteínas, llamadas oligoméricas, que están formadas por varias cadenas polipeptídicas, por lo que los términos cadena polipeptídica y proteína no pueden considerarse sinónimos en todos los casos.
Aunque en los sucesivo nos ocuparemos fundamentalmente de proteínas formadas por centenares de residuos de aminoácidos, es conveniente resaltar que algunos péptidos cortos presentan actividades biológicas importantes. Entre ellos cabe citar algunas hormonas como la oxitocina (nueve residuos aminoácidos), que estimula las contracciones del útero durante el parto, o la bradiquinina (nueve residuos), que inhibe la inflamación de los tejidos. También son dignas de mención las encefalinas, péptidos cortos sintetizados en el sistema nervioso central que actúan sobre el cerebro produciendo analgesia (eliminación del dolor). Los venenos extremadamente tóxicos producidos por algunas setas como Amanita phaloides también son péptidos, al igual que muchos antibióticos.
7.-PROTEÍNAS: CONFORMACIÓN TRIDIMENSIONAL.
Las proteínas, como ya se dijo anteriormente, no son polímeros al azar de longitud indefinida, sino que cada una de ellas tiene una determinada composición en aminoácidos y estos están ordenados en una determinada secuencia. Hay que añadir a ello que en las células vivas las cadenas polipeptídicas de las proteínas no se encuentran extendidas ni plegadas al azar adoptando estructuras caprichosas o variables, sino que cada una de ellas se encuentra plegada de un modo característico, que es igual para todas las moléculas de una misma proteína, y que recibe el nombre de estructura o conformación tridimensional nativa de la proteína. Una clara evidencia en favor de esta idea la constituye el hecho de que las proteínas puedan cristalizar, ya que la disposición ordenada de la materia cristalina sólo es posible cuando las unidades moleculares individuales que componen el cristal son idénticas. Desde que en 1926 James Sumner consiguió obtener cristales del enzima ureasa, centenares de proteínas han sido obtenidas en estado cristalino.
El plegamiento de una cadena polipeptídica se realiza mediante rotaciones de los enlaces simples del esqueleto. En principio, los sustituyentes de los átomos que se encuentran a ambos lados de un enlace simple pueden adoptar infinitas posiciones (conformaciones) mediante simples rotaciones de dicho enlace. Dado que el esqueleto de una cadena polipeptídica consta de centenares de enlaces simples, cabría esperar que dicha cadena pudiera adoptar un número elevadísimo de conformaciones diferentes. Sin embargo, existen una serie de restricciones a la libertad de giro de estos enlaces (la mayoría de ellas derivadas de la interacción de la cadena polipeptídica con las moléculas de agua que la rodean) las cuales determinan que sólo sea posible una conformación tridimensional estable.
La conformación tridimensional de una proteína es un hecho biológico de una gran complejidad: existen distintos niveles de plegamiento que se superponen unos a otros. Debido a ello, para sistematizar el conocimiento acerca de este fenómeno, se establecen una serie de niveles dentro de la estructura de la proteína que se conocen como estructuras primaria, secundaria, terciaria y cuaternaria (Figura 8.6). Los continuos avances en la comprensión de la estructura y el plegamiento de las proteínas han hecho necesaria en los últimos años la definición de dos niveles estructurales adicionales: la estructura supersecundaria y el dominio.
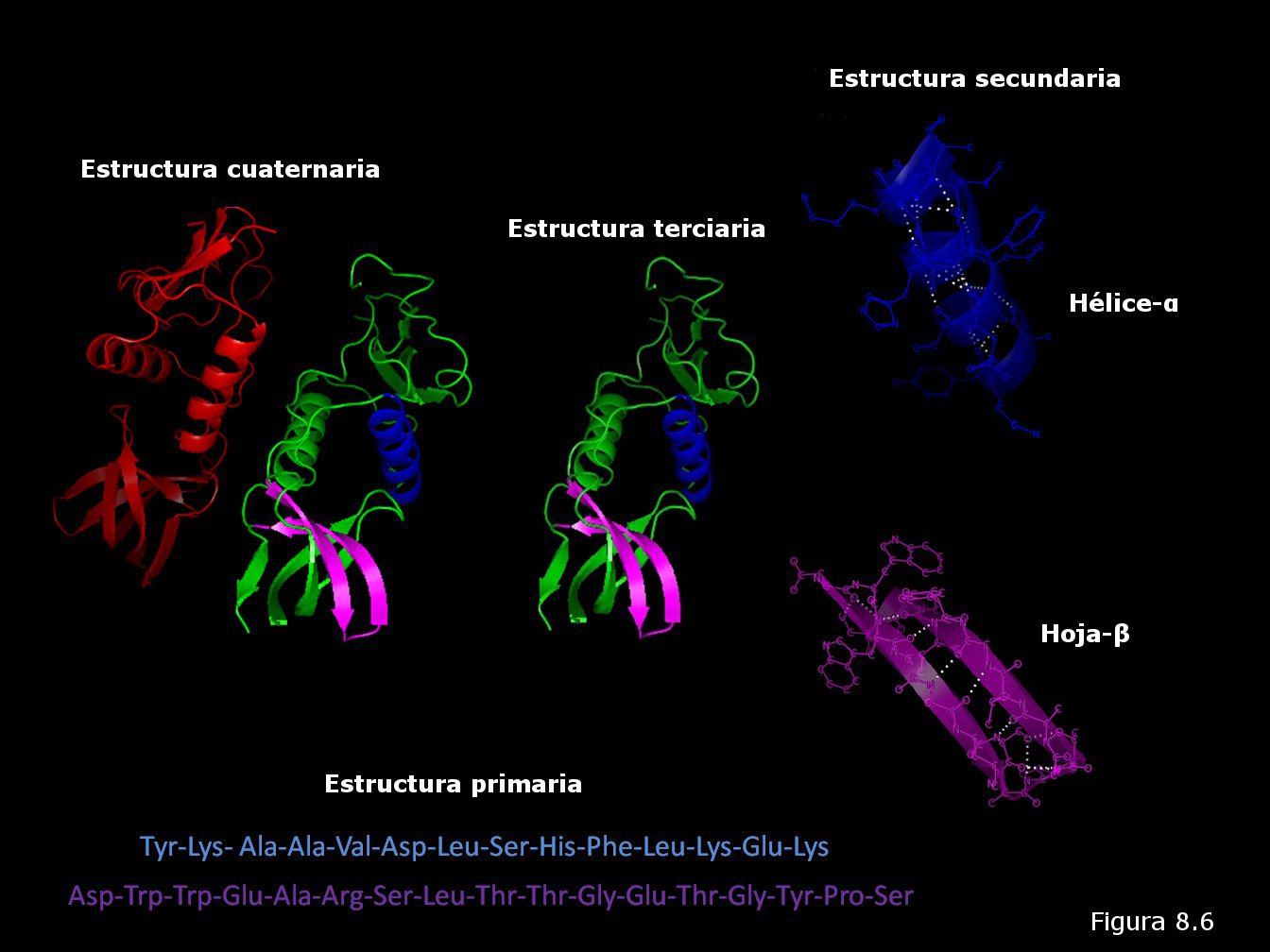
7.1.-ESTRUCTURA PRIMARIA.
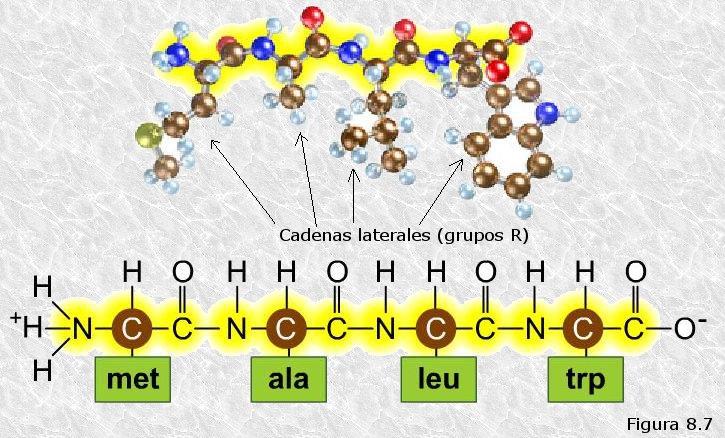
La estructura primaria de una proteína es su secuencia de aminoácidos, es decir, vendría especificada por los aminoácidos que la forman y el orden de colocación de los mismos a lo largo de la cadena polipeptídica. La secuencia de aminoácidos de una proteína se escribe empezando por el extremo amino terminal y finalizando por el carboxi-terminal.
Si analizamos en detalle la estructura primaria de una proteína (ver Figura 8.7) observaremos, dado que el enlace peptídico implica a los grupos amino y carboxilo de cada aminoácido, y que éstos están unidos a su vez al mismo átomo de carbono (Cα), el esqueleto de la cadena polipeptídica es una sucesión monótona de estos tres tipos de enlace:
C α ------- C carboxílico
C carbox --- N amino(enlace peptídico)
N amino ---- C α
También observamos que las cadenas laterales o grupos R de los distintos restos aminoácidos, que no están implicadas en el enlace peptídico, surgen lateralmente hacia afuera de este esqueleto monótono (ver Figura 8.7).
Los estudios realizados acerca de la estructura primaria de proteínas procedentes de diferentes especies de seres vivos revelan que aquellas proteínas que desempeñan funciones similares en diferentes especies tienen secuencias de aminoácidos parecidas entre sí. Por otra parte, se ha comprobado que cuanto más emparentadas evolutivamente estén dos especies mayor es el grado de similitud entre las secuencias de aminoácidos de sus proteínas homólogas. Estos datos sugieren que debe existir algún tipo de relación entre la secuencia de aminoácidos y la función de las proteínas.
7.2.-ESTRUCTURA SECUNDARIA.
La estructura secundaria de una proteína es el modo característico de plegarse la misma a lo largo de un eje. Es el primer nivel de plegamiento, en el que los distintos restos de aminoácidos se disponen de un modo ordenado y repetitivo siguiendo una determinada dirección. En las proteínas fibrosas (aquéllas cuyas cadenas polipeptídicas están ordenadas formando largos filamentos u hojas planas) las estructuras primaria y secundaria especifican completamente la conformación tridimensional; estas proteínas no presentan por lo tanto niveles superiores de complejidad
Fue precisamente en las proteínas fibrosas, dada su mayor simplicidad estructural, donde fue estudiada inicialmente la estructura secundaria; particularmente en dos tipos de proteínas de origen animal muy abundantes: las queratinas y los colágenos. Ambas son proteínas insolubles que desempeñan importantes funciones de tipo estructural en los animales superiores. Existen dos tipos de queratinas de diferente dureza y consistencia, las α-queratinas (por ejemplo las que abundan en el pelo o en las uñas) y las β-queratinas (telas de araña, seda, etc.).
El análisis de la estructura secundaria de las proteínas fibrosas
fue abordado
 inicialmente mediante la técnica de difracción de
rayos X (basada en la capacidad de los átomos de difractar los RX en función de su tamaño). Esta técnica es aplicable al análisis
de estructuras cristalinas, sin embargo, la microscopía electrónica
reveló que las proteínas fibrosas presentaban estructuras
repetitivas que eran susceptibles de análisis mediante esta técnica.
inicialmente mediante la técnica de difracción de
rayos X (basada en la capacidad de los átomos de difractar los RX en función de su tamaño). Esta técnica es aplicable al análisis
de estructuras cristalinas, sin embargo, la microscopía electrónica
reveló que las proteínas fibrosas presentaban estructuras
repetitivas que eran susceptibles de análisis mediante esta técnica.
Los primeros análisis de difracción de rayos X de las queratinas, realizados por Willian Atsbury (Figura 8.8) en la década de los años 30, proporcionaron datos acerca de estructuras que se repetían con una periodicidad fija a lo largo de sus cadenas polipeptídicas, siendo estas periodicidades diferentes según se tratase de α o de β-queratinas. Dado que las cadenas polipeptídicas extendidas no presentan estructuras repetitivas que puedan dar lugar a estas periodicidades, se concluyó que dichas cadenas debían encontrarse plegadas de un modo regular que era diferente en cada tipo de queratinas. Pocos años más tarde, L. Pauling y R. Corey (Figura 8.9), dos investigadores norteamericanos, obtuvieron con gran precisión la longitud de estas periodicidades (0,56 nm en las α y 0,70 nm en las β-queratinas).
Por otra parte, aplicando la técnica DRX a pequeños péptidos (dos o
tres residuos aminoácidos) en estado cristalino Pauling y Corey
pudieron conocer la estructura íntima del enlace peptídico.
Observaron que este enlace era ligeramente más corto de lo que sería
un enlace simple C-N, lo que les permitió deducir que poseía un
carácter parcial de doble enlace. Ello es debido a que el nitrógeno
del grupo peptídico posee un orbital vacante que le permite
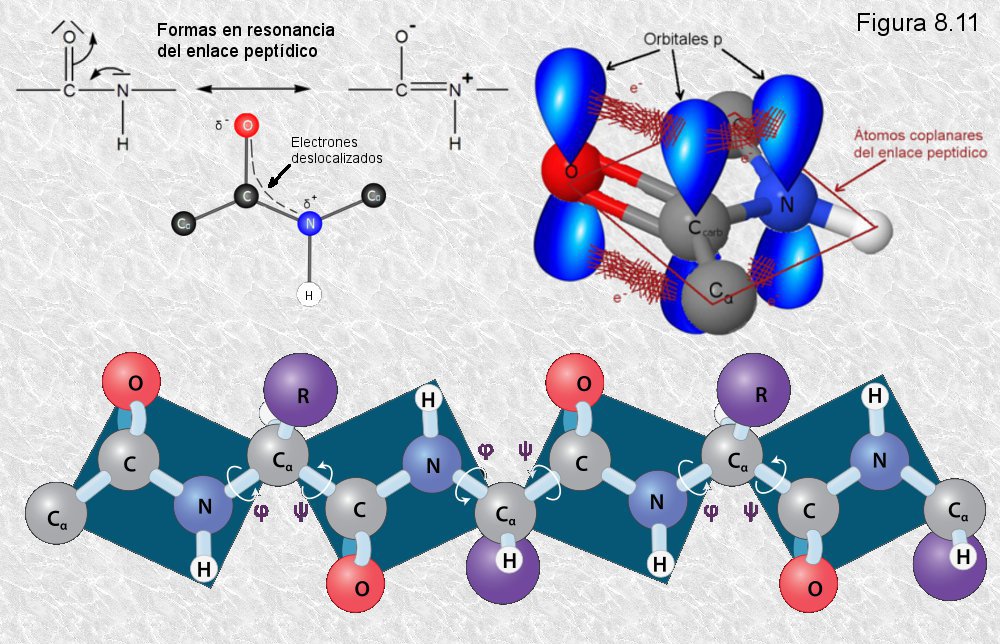
compartir en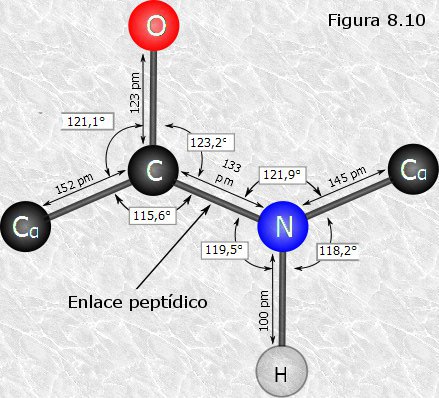 resonancia un par de electrones del doble enlace
C-O. El carácter parcial de doble enlace impide que el enlace
peptídico pueda girar sobre sí mismo; los cuatro átomos del grupo
peptídico son coplanares, estando el oxígeno y el hidrógeno en
posición trans (Figura
8.10). Esta falta de libertad de giro supone una primera
restricción en el número de conformaciones posibles de la cadena
polipeptídica, que estaría entonces constituida por una serie de
planos rígidos formados por los diferentes grupos peptídicos, los
cuales podrían adoptar diferentes posiciones unos con respecto a
otros mediante giros de los enlaces sencillos que flanquean cada uno
de estos planos (ver
Figura 8.11).
resonancia un par de electrones del doble enlace
C-O. El carácter parcial de doble enlace impide que el enlace
peptídico pueda girar sobre sí mismo; los cuatro átomos del grupo
peptídico son coplanares, estando el oxígeno y el hidrógeno en
posición trans (Figura
8.10). Esta falta de libertad de giro supone una primera
restricción en el número de conformaciones posibles de la cadena
polipeptídica, que estaría entonces constituida por una serie de
planos rígidos formados por los diferentes grupos peptídicos, los
cuales podrían adoptar diferentes posiciones unos con respecto a
otros mediante giros de los enlaces sencillos que flanquean cada uno
de estos planos (ver
Figura 8.11).
Pauling y Corey construyeron modelos moleculares de gran precisión (con bolas y varillas) hasta que encontraron unos que encajaban con los datos experimentales, es decir, hasta que encontraron modelos que, respetando las restricciones de giro del enlace peptídico, explicaban las periodicidades obtenidas. A la vista de estos modelos pudieron observar (suponemos que con gran regocijo) que no sólo eran posibles, sino que, de ser reales, presentarían una gran estabilidad, ya que todos los grupos peptídicos del esqueleto quedaban colocados en la relación geométrica adecuada para poder establecer puentes de hidrógeno entre ellos, circunstancia esta que proporcionaría una gran estabilidad a la estructura.
Los modelos encontrados fueron denominados respectivamente hélice α (que es la estructura secundaria de las α-queratinas) y conformación β (que es la estructura secundaria de las β-queratinas). Con posterioridad se descubrió la estructura secundaria del colágeno, la cual se denominó hélice del colágeno.
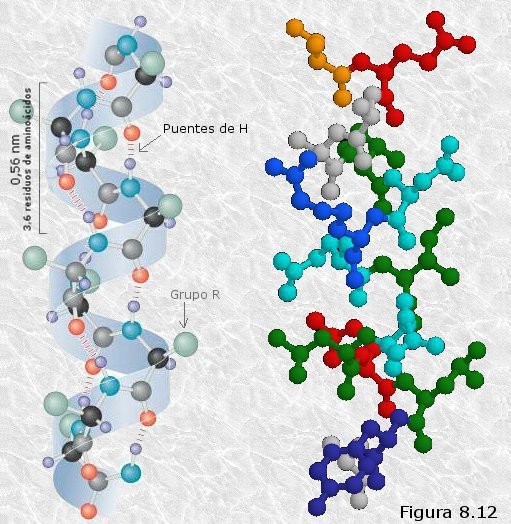 En la hélice-α (ver
Figura 8.12) el esqueleto de la cadena polipeptídica se encuentra
arrollado de manera compacta alrededor del eje longitudinal de la
molécula, y los grupos R de los distintos restos aminoácidos
sobresalen de esta estructura helicoidal, que tiene forma de
escalera de caracol. Cada giro de la hélice abarca 3,6 residuos
aminoácidos, ocupando unos 0,56 nm del eje longitudinal, lo que se
corresponde con la periodicidad observada por DRX. El rasgo más
sobresaliente de esta estructura es que todos los grupos peptídicos
de los diferentes restos aminoácidos quedan enfrentados en la
relación geométrica adecuada para formar puentes de hidrógeno entre
sí; estos puentes se establecen entre el oxígeno del grupo carboxilo
de cada residuo aminoácido y el hidrógeno del grupo amino que se
encuentra cuatro residuos más allá en dirección carboxi-terminal
(algo más de una vuelta completa de hélice). Así, cada vuelta
sucesiva de la hélice α se mantiene unida a las vueltas adyacentes
mediante varios puentes de hidrógeno intracatenarios que,
actuando cooperativamente, proporcionan a la estructura una
considerable estabilidad.
En la hélice-α (ver
Figura 8.12) el esqueleto de la cadena polipeptídica se encuentra
arrollado de manera compacta alrededor del eje longitudinal de la
molécula, y los grupos R de los distintos restos aminoácidos
sobresalen de esta estructura helicoidal, que tiene forma de
escalera de caracol. Cada giro de la hélice abarca 3,6 residuos
aminoácidos, ocupando unos 0,56 nm del eje longitudinal, lo que se
corresponde con la periodicidad observada por DRX. El rasgo más
sobresaliente de esta estructura es que todos los grupos peptídicos
de los diferentes restos aminoácidos quedan enfrentados en la
relación geométrica adecuada para formar puentes de hidrógeno entre
sí; estos puentes se establecen entre el oxígeno del grupo carboxilo
de cada residuo aminoácido y el hidrógeno del grupo amino que se
encuentra cuatro residuos más allá en dirección carboxi-terminal
(algo más de una vuelta completa de hélice). Así, cada vuelta
sucesiva de la hélice α se mantiene unida a las vueltas adyacentes
mediante varios puentes de hidrógeno intracatenarios que,
actuando cooperativamente, proporcionan a la estructura una
considerable estabilidad.
En la conformación β, también llamada hoja plegada, el
esqueleto de la cadena polipeptídica se dispone en zig-zag
con los grupos R de los distintos aminoácidos proyectándose
alternativamente a uno y otro lado de dicho esqueleto (ver
Figura 8.13). Cada zig-zag representa 0,70 nm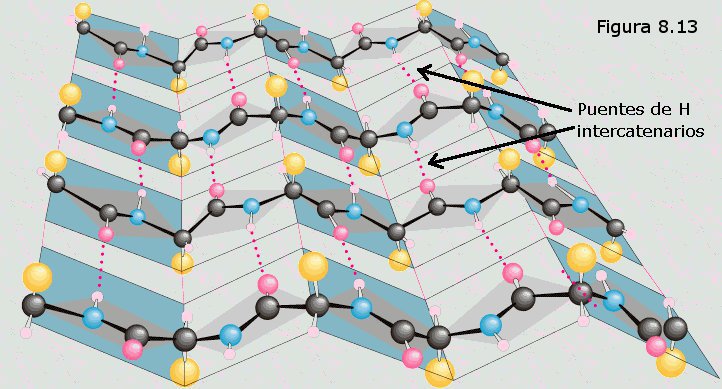 de longitud
de la cadena, coincidiendo con la periodicidad observada por DRX.
Muchas de estas cadenas colocadas paralelamente unas a otras forman
una estructura que recuerda a una hoja de papel plegada, en la que
los grupos R de los aminoácidos se encuentran sobresaliendo por
ambas caras de dicha hoja. En este caso, los grupos peptídicos de
los diferentes restos aminoácidos establecen puentes de hidrógeno
con los de las cadenas vecinas (puentes de hidrógeno
intercatenarios).
de longitud
de la cadena, coincidiendo con la periodicidad observada por DRX.
Muchas de estas cadenas colocadas paralelamente unas a otras forman
una estructura que recuerda a una hoja de papel plegada, en la que
los grupos R de los aminoácidos se encuentran sobresaliendo por
ambas caras de dicha hoja. En este caso, los grupos peptídicos de
los diferentes restos aminoácidos establecen puentes de hidrógeno
con los de las cadenas vecinas (puentes de hidrógeno
intercatenarios).
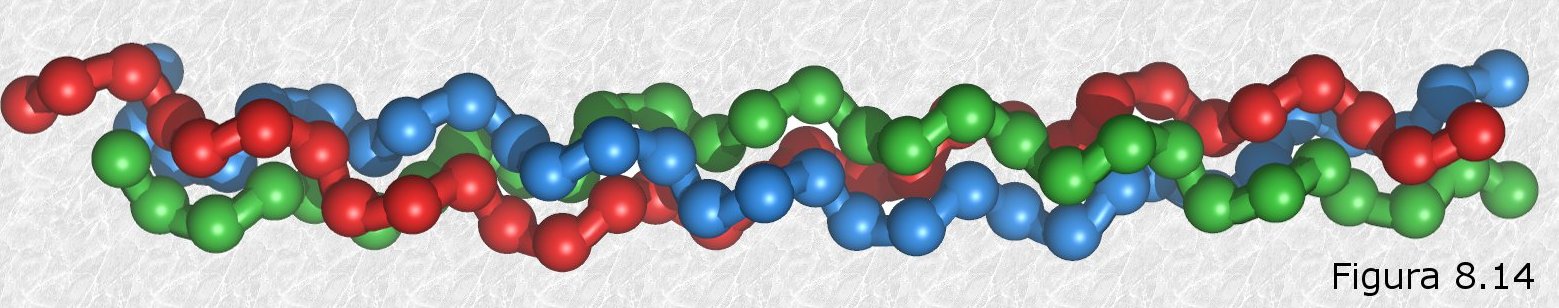
La hélice del colágeno (Figura 8.14) es un tipo de estructura secundaria que sólo aparece en esta proteína. Se trata de un arrollamiento helicoidal con tres residuos aminoácidos por vuelta en el que la cadena polipeptídica se encuentra más extendida que en la hélice α. Tres de estas hélices se encuentran a su vez arrolladas en una estructura superhelicoidal que da lugar a la molécula de tropocolágeno, que es la unidad que se repite a lo largo de las fibras de colágeno.
La hélice-α y la conformación β son las estructuras secundarias más
frecuentes no sólo en la proteínas fibrosas sino en todo tipo de
proteínas. Existen otros tipos de estructura secundaria cuya
presencia se encuentra limitada a algunas proteínas especializadas.
En las proteínas globulares se han descubierto estructuras secundarias características de los puntos en que la cadena polipeptídica cambia abruptamente de dirección. Se les ha denominado giros o codos. El más extendido es el codo β, que consta de cuatro residuos aminoácidos formando un bucle cerrado con diferentes grupos peptídicos unidos por puente de hidrógeno (Figura 8.15).
Ahora bien, ¿qué es lo que determina que una cadena polipeptídica adopte una u otra de estas posibles estructuras secundarias conocidas? Los estudios realizados acerca de la estructura de cadenas polipeptídicas formadas por un solo tipo aminoácido (poliaminoácidos), así como diversas consideraciones teóricas (basadas en el tamaño o carga eléctrica de los grupos R de los distintos aminoácidos de una cadena), llevaron a la conclusión de que es la secuencia de aminoácidos, es decir, la estructura primaria, lo que determina el modo en que una cadena polipeptídica ha de plegarse a lo largo de un eje, es decir, su estructura secundaria. Es la naturaleza y posición de los grupos R a lo largo de la cadena, es decir, su secuencia, lo que propicia o impide el plegamiento según uno u otro modelo.
7.3.-ESTRUCTURA TERCIARIA.
 Existen proteínas cuya conformación tridimensional no puede
especificarse totalmente considerando sólo sus estructuras primaria
y secundaria. Son las llamadas proteínas globulares cuyas cadenas
polipeptídicas se hallan plegadas de un modo complejo formando
arrollamientos globulares compactos que tienden a adoptar una forma
aproximadamente esférica. La proteínas globulares son generalmente
solubles en agua y desempeñan un gran número de funciones biológicas
(por ejemplo los enzimas son proteínas globulares). Se conoce como
estructura terciaria el modo característico de plegarse una
cadena polipeptídica para formar un arrollamiento globular compacto.
Existen proteínas cuya conformación tridimensional no puede
especificarse totalmente considerando sólo sus estructuras primaria
y secundaria. Son las llamadas proteínas globulares cuyas cadenas
polipeptídicas se hallan plegadas de un modo complejo formando
arrollamientos globulares compactos que tienden a adoptar una forma
aproximadamente esférica. La proteínas globulares son generalmente
solubles en agua y desempeñan un gran número de funciones biológicas
(por ejemplo los enzimas son proteínas globulares). Se conoce como
estructura terciaria el modo característico de plegarse una
cadena polipeptídica para formar un arrollamiento globular compacto.
El estudio de la estructura terciaria de las proteínas globulares se
abordó también mediante la aplicación de la técnica de difracción de
rayos X. Un paso previo necesario fue la obtención de proteínas
globulares en estado cristalino muy puro a partir de disoluciones,
ya que la técnica DRX sólo se puede aplicar a estructuras
cristalinas o "paracristalinas" (como las proteínas fibrosas). Una
vez solventado este problema pudo conocerse la estructura terciaria
de algunas proteínas globulares (tras años de trabajo para conseguir
interpretar los complejos difractogramas de RX que estas proteínas
producían. Véase la
Figura 8.16).
Los cristalógrafos ingleses John Kendrew y Max Perutz (Figura 8.16b)
obtuvieron grandes éxitos en la elucidación de la estructura
tridimensional de proteínas globulares. La primera proteína cuya estructura terciaria fue
conocida fue la mioglobina (una proteína que transporta oxígeno en
el músculo), concretamente la del cachalote (ver
Figura 8.17). En ella se pueden apreciar ocho segmentos
rectilíneos con estructura secundaria en hélice α separados por
curvaturas sin estructura secundaria aparente. Alrededor del 70% de
la cadena polipeptídica se encuentra en las regiones plegadas en
hélice α. La molécula es muy compacta, sin apenas espacio para
moléculas de agua en su interior. Los grupos R de residuos
aminoácidos con carácter polar o iónico se proyectan hacia la
periferia de la molécula, mientras que los de carácter no polar se
encuentran enterrados en el interior de la misma, aislados del
contacto con el agua. La estructura se encuentra estabilizada por
diferentes tipos de interacciones débiles entre los grupos R de
diferentes aminoácidos; estas interacciones son de largo alcance,
afectando a grupos R de residuos aminoácidos que pueden ocupar
posiciones muy alejadas en la cadena polipeptídica.
proteínas
producían. Véase la
Figura 8.16).
Los cristalógrafos ingleses John Kendrew y Max Perutz (Figura 8.16b)
obtuvieron grandes éxitos en la elucidación de la estructura
tridimensional de proteínas globulares. La primera proteína cuya estructura terciaria fue
conocida fue la mioglobina (una proteína que transporta oxígeno en
el músculo), concretamente la del cachalote (ver
Figura 8.17). En ella se pueden apreciar ocho segmentos
rectilíneos con estructura secundaria en hélice α separados por
curvaturas sin estructura secundaria aparente. Alrededor del 70% de
la cadena polipeptídica se encuentra en las regiones plegadas en
hélice α. La molécula es muy compacta, sin apenas espacio para
moléculas de agua en su interior. Los grupos R de residuos
aminoácidos con carácter polar o iónico se proyectan hacia la
periferia de la molécula, mientras que los de carácter no polar se
encuentran enterrados en el interior de la misma, aislados del
contacto con el agua. La estructura se encuentra estabilizada por
diferentes tipos de interacciones débiles entre los grupos R de
diferentes aminoácidos; estas interacciones son de largo alcance,
afectando a grupos R de residuos aminoácidos que pueden ocupar
posiciones muy alejadas en la cadena polipeptídica.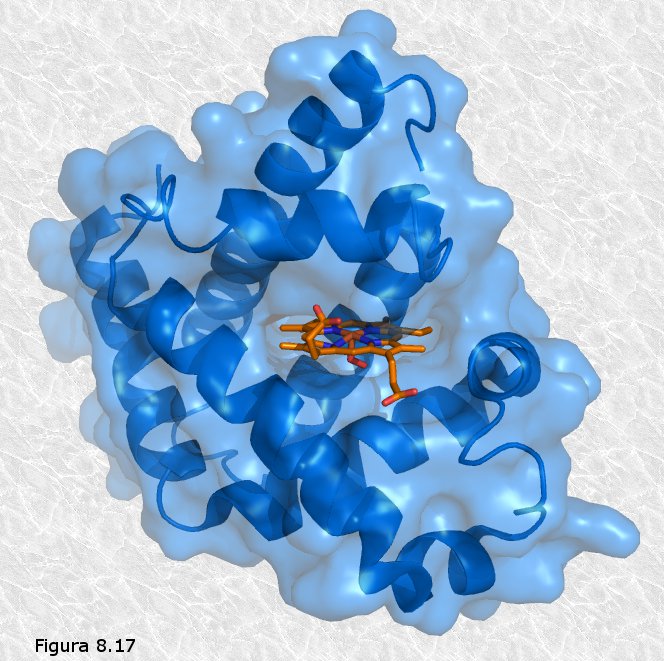
En los últimos años se ha podido descifrar la estructura terciaria de miles de proteínas globulares. De estos estudios se deduce que el caso de la mioglobina representa sólo una de las múltiples posibilidades de plegamiento de una proteína globular. La variedad de estructuras terciarias posibles es inmensa. Sin embargo se pueden hacer algunas generalizaciones interesantes. En todas ellas...
a) La cadena polipeptídica está plegada de un modo muy compacto, sin apenas espacio para moléculas de agua en el interior del plegamiento.
b)
Existen tramos rectilíneos que presentan estructura secundaria en
hélice-α o en conformación β; en la mayoría de las proteínas
estudiadas coexisten zonas con
uno y otro tipo de estructura
(Figura 8.18). Estos tramos están separados por
curvaturas sin estructura secundaria aparente en unos casos o por
codos β en otros. Las cantidades relativas que representan los
diferentes tipos de estructura secundaria varían considerablemente
de unas proteínas a otras.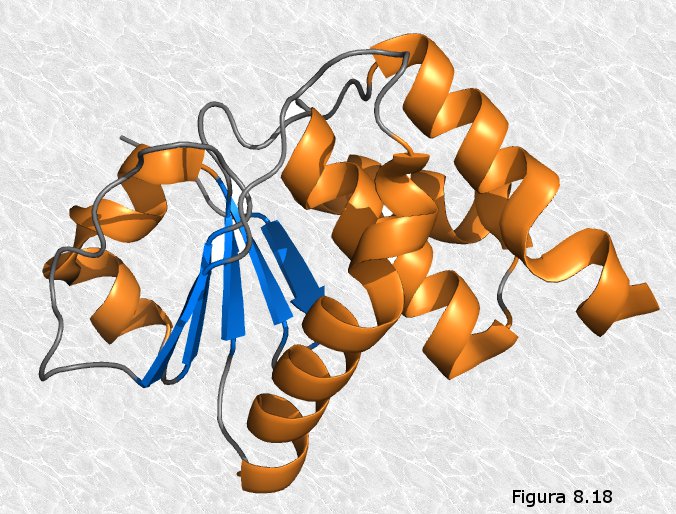
c) Se han detectado agrupamientos estables de estructuras secundarias que dan lugar a motivos estructurales que se repiten en multitud de proteínas diferentes. Entre estos agrupamientos, denominados estructuras supersecundarias, cabe citar el "barril α/β", la "silla β", el "haz de cuatro hélices", el "lazo βαβ" o el "sandwich ββ".
d) En algunas proteínas se han detectado dos o más regiones globulares densamente empaquetadas que se hallan conectadas entre sí por un corto tramo de cadena polipeptídica extendida o plegada en hélice α. Estas regiones globulares, denominadas dominios, presentan una gran estabilidad, y aparecen repetidas en muchas proteínas diferentes.
e) Los restos de aminoácidos con grupos R polares o con carga se proyectan hacia el exterior de la estructura, expuestos al contacto con las moléculas de agua.
f) Los restos de aminoácidos con grupos R no polares (hidrófobos) se encuentran en el interior de la estructura, aislados del contacto con el agua y ejerciendo interacciones hidrofóbicas entre sí.
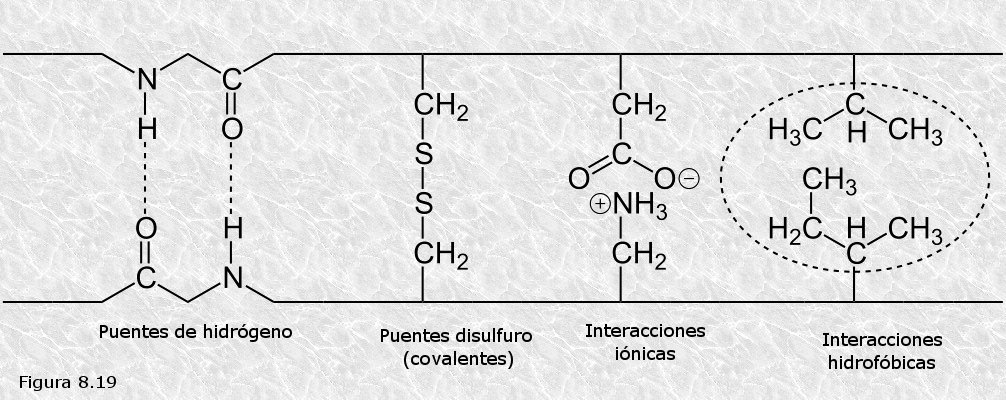
Por otra parte se observó que en todas las proteínas estudiadas existe una serie de fuerzas intramoleculares que tienden a estabilizar la estructura terciaria (Figura 8.19). Estas fuerzas son de dos tipos: a) enlaces covalentes (puentes disulfuro entre los grupos -SH de los restos de cisteína); b) interacciones débiles entre los grupos R de distintos aminoácidos que ocupan posiciones muy distantes a lo largo de la cadena polipeptídica (puentes de hidrógeno, interacciones iónicas, fuerzas de Van der Waals).
A la vista de estos datos se llegó a la conclusión de que es la naturaleza (polar o no polar) de los distintos grupos R, las posibilidades de formación de interacciones débiles o covalentes entre los mismos, y su posición en la cadena polipeptídica, es decir, la secuencia de aminoácidos lo que determina el hecho de que ésta adopte una u otra disposición en el espacio, o lo que es lo mismo, una determinada estructura terciaria. Hay que tener en cuenta que en su estado nativo las moléculas proteicas se encuentran en el seno del agua y que, por lo tanto, el plegamiento de la cadena polipeptídica será una respuesta a la interacción de los distintos grupos R (polares o no polares) con las moléculas de agua; además, la posibilidad de que se establezcan interacciones que estabilicen la estructura entre los distintos grupos R a lo largo de la cadena polipeptídica también dependerá de la naturaleza y posición de los mismos en la cadena. En la actualidad todo parece indicar que las interacciones hidrofóbicas entre los grupos R no polares enterrados en el interior de la estructura constituyen la verdadera fuerza directriz del plegamiento de una cadena polipeptídica, contribuyendo los demás tipos de interacciones débiles y covalentes a su mayor estabilidad.
Vemos, pues, que, al igual que sucede con la estructura secundaria, la estructura primaria determina la estructura terciaria de las proteínas globulares.
7.4.-ESTRUCTURA CUATERNARIA.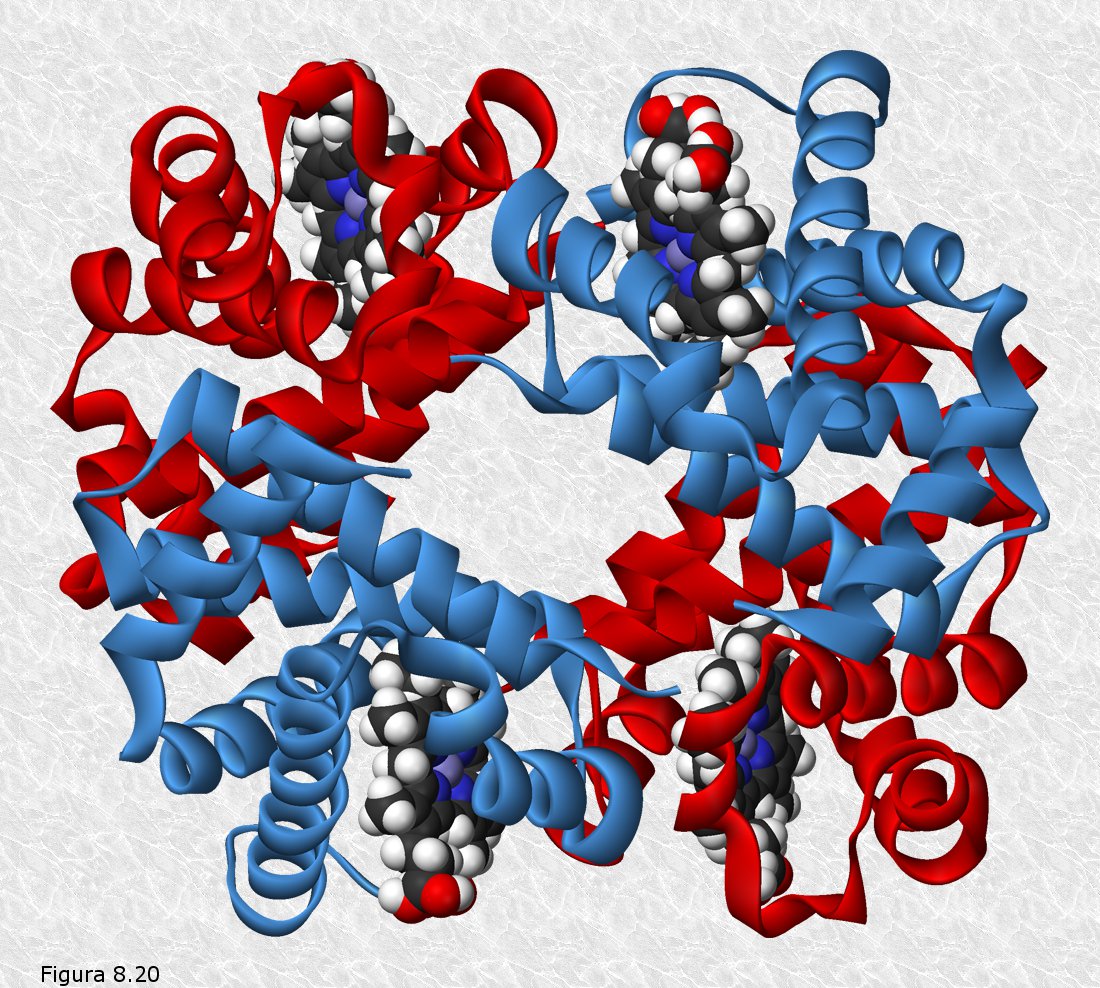
Existen proteínas que están formadas por varias cadenas polipeptídicas: son las llamadas proteínas oligoméricas. En ellas, la proteína completa (oligómero) está formada por un número variable de subunidades o protómeros. Los oligómeros pueden ser dímeros, trímeros, tetrámeros, pentámeros, hexámeros...., según estén formados por 2, 3, 4, 5, 6.... protómeros. Los oligómeros más frecuentes están formados por un número par de cadenas polipeptídicas.
En estas proteínas las distintas subunidades están asociadas de un modo característico al que llamamos estructura cuaternaria.
El estudio de la estructura cuaternaria de las proteínas oligoméricas también fue abordado mediante la aplicación de la técnica DRX tras la obtención de las mismas en estado cristalino puro. En este caso la interpretación de los difractogramas de RX resultó tan compleja que algunos cristalógrafos de proteínas emplearon en este esfuerzo hasta 25 años de trabajo antes de poder publicar resultados.
La primera proteína cuya estructura cuaternaria fue conocida (Figura 8.20) fue la hemoglobina humana (la proteína encargada de transportar el oxígeno en la sangre).
También se pueden hacer algunas generalizaciones acerca de la estructura cuaternaria de algunas proteínas oligoméricas conocidas. En todas ellas.....
a) Cada una de las subunidades o protómeros presenta una estructura terciaria determinada con rasgos similares a los de las proteínas globulares formadas por una sola cadena polipeptídica.
b) La estructura terciaria de las diferentes subunidades de una proteína oligomérica es muy semejante a la de proteínas globulares que desempeñan la misma o parecida función (la estructura terciaria de cada una de las subunidades de la hemoglobina es casi idéntica a la estructura terciaria de la mioglobina. Ambas proteínas desempeñan la función de transportar oxígeno, una en la sangre, la otra en el músculo. Se percibe pues una clara relación entre estructura y función.
c) Las distintas subunidades se encuentran asociadas de un modo característico, estableciéndose entre ellas puntos de contacto que son los mismos para todas las moléculas de una misma proteína. Estos puntos de contacto se ven estabilizados por interacciones débiles (puentes de hidrógeno, fuerzas de Van der Waals, interacciones iónicas) entre los grupos R de determinados aminoácidos.
A la vista de estos resultados se dedujo que también en este caso es la naturaleza y posición de los grupos R de los distintos aminoácidos en las diferentes subunidades la que determina cuáles han de ser los puntos de contacto entre las mismas, y, por lo tanto, el modo característico de asociarse unas con otras, es decir, la estructura cuaternaria; los puntos de contacto vendrán dados por las posibilidades de formación de interacciones débiles del tipo de las citadas y éstas a su vez de la naturaleza y posición de los distintos grupos R.
Deducimos, pues, que es la estructura primaria de las distintas subunidades la que determina la estructura cuaternaria de una proteína oligomérica.
Como conclusión podemos afirmar que la secuencia de aminoácidos (estructura primaria) contiene la información necesaria y suficiente para determinar la conformación tridimensional de una proteína a sus diferentes niveles de complejidad (estructuras secundaria, terciaria y cuaternaria).
8.-PROTEÍNAS. RELACIÓN ESTRUCTURA-FUNCIÓN.
Las proteínas son las macromoléculas más versátiles de cuantas existen en la materia viva: desempeñan un elevado número de funciones biológicas diferentes. Cada proteína está especializada en llevar a cabo una determinada función.
Entre las funciones de las proteínas cabe destacar las siguientes: catalíticas, estructurales, de transporte, nutrientes y de reserva, contráctiles o mótiles, de defensa, reguladoras del metabolismo, y otras muchas que determinadas proteínas desempeñan en organismos concretos.
La función de una proteína depende de la interacción de la misma con una
molécula a la que llamamos ligando (en el caso particular de los
enzimas el ligando recibe el nombre de sustrato). El ligando es
específico de cada proteína. A su vez, la interacción entre proteína y
ligando reside en un principio de complementariedad estructural:
el ligando debe encajar en un hueco existente en la superficie de la
proteína (el centro activo) tal y como lo haría una llave en una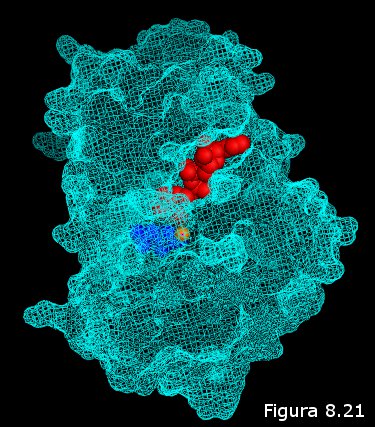 cerradura (ver
Figura 8.21). Sólo aquel o aquellos ligandos capaces de acoplarse en
el centro activo de la proteína serán susceptibles de interactuar con
ella. Hay que tener en cuenta que este acoplamiento no es meramente
espacial, sino que la proteína "ve" en su ligando, además de la forma,
la distribución de cargas eléctricas, sus distintos grupos funcionales,
y, en general, las posibilidades de establecer interacciones débiles con
él a través de los grupos R de los aminoácidos que rodean el centro
activo (el ligando "atraca" en el centro activo como lo haría un barco
en un muelle, se establecen entre ambos "amarras" en forma de
interacciones débiles que hacen más estable la asociación).
cerradura (ver
Figura 8.21). Sólo aquel o aquellos ligandos capaces de acoplarse en
el centro activo de la proteína serán susceptibles de interactuar con
ella. Hay que tener en cuenta que este acoplamiento no es meramente
espacial, sino que la proteína "ve" en su ligando, además de la forma,
la distribución de cargas eléctricas, sus distintos grupos funcionales,
y, en general, las posibilidades de establecer interacciones débiles con
él a través de los grupos R de los aminoácidos que rodean el centro
activo (el ligando "atraca" en el centro activo como lo haría un barco
en un muelle, se establecen entre ambos "amarras" en forma de
interacciones débiles que hacen más estable la asociación).
De lo anteriormente expuesto es fácil deducir que para que una proteína desempeñe su función biológica debe permanecer intacta su conformación tridimensional nativa. Si se pierde dicha conformación, y por lo tanto se altera la estructura del centro activo, ya no habrá acoplamiento entre proteína y ligando (no se "reconocerán") y la interacción entre ambos, de la que depende la función, ya no tendrá lugar. Como corolario de este razonamiento podemos afirmar que la función biológica de una proteína depende de su conformación tridimensional.
En resumen, la secuencia de aminoácidos de una proteína determina su conformación tridimensional, y ésta, a su vez, su función biológica.
9.-DESNATURALIZACIÓN DE LAS PROTEÍNAS.
Se entiende por desnaturalización de una proteína la pérdida de la conformación tridimensional nativa de la misma, pérdida que suele ir acompañada de un descenso en la solubilidad (las cadenas polipeptídicas de la proteína desnaturalizada se agregan unas a otras y forman un precipitado que se separa de la disolución). Durante el proceso de desnaturalización se rompen las interacciones débiles que mantienen estable la conformación pero se mantienen los enlaces covalentes del esqueleto polipeptídico, es decir, se pierden las estructuras secundaria, terciaria y, en su caso, cuaternaria, pero permanece intacta la secuencia de aminoácidos.
La desnaturalización puede ser provocada por diferentes causas o agentes desnaturalizantes de tipo físico o químico. Destacaremos dos de ellos: uno físico (aumento de temperatura) y otro químico (alteración del pH).
a) Aumento de temperatura.- Los aumentos de temperatura provocan una mayor agitación molecular que hace que las interacciones débiles que mantienen estable la conformación de la proteína terminen por ceder con la consiguiente desnaturalización.
b) Alteración del pH.- Estas alteraciones causan variación en el grado de ionización de distintos grupos funcionales (carboxilo, amino, hidroxilo, etc.) implicados en interacciones débiles que estabilizan la conformación. Estas variaciones provocan la rotura de dichas interacciones (sobre todo enlaces iónicos y también puentes de hidrógeno) y por lo tanto la desnaturalización (debido a ello son tan importantes los tampones que mantienen estable el pH de los fluidos biológicos).
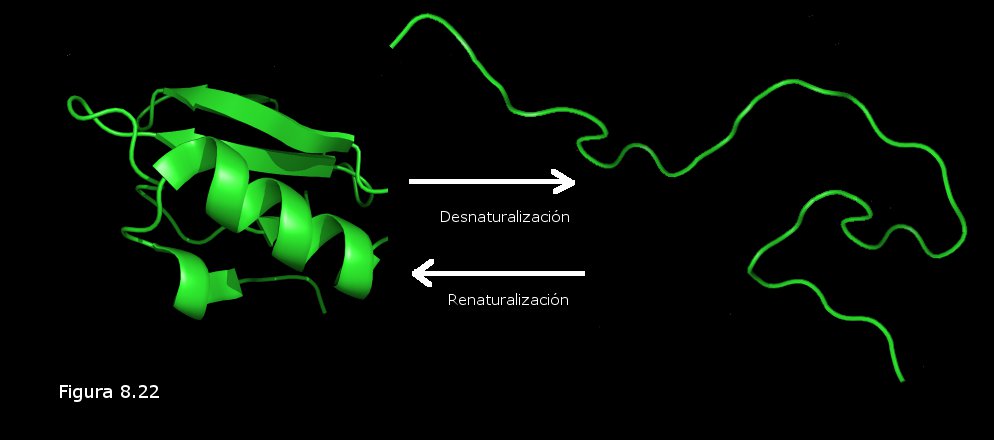
El proceso de desnaturalización, si se lleva a cabo en condiciones suaves (variaciones moderadas y graduales de temperatura o pH), es reversible: la proteína puede recuperar su conformación tridimensional nativa si se restituyen las condiciones iniciales. Este proceso recibe el nombre de renaturalización. En la Figura 8.22 se ilustra el proceso de desnaturalización reversible de una cadena polipeptídica. Se ha comprobado en multitud de experimentos que el proceso de renaturalización conlleva una recuperación de la función biológica de la proteína (que se había perdido durante la desnaturalización), lo cual constituye una prueba irrefutable de la singular relación existente entre la secuencia de aminoácidos, la conformación tridimensional, y la función biológica de una proteína: la secuencia de aminoácidos, que es lo único que permanece al final del proceso de desnaturalización, contiene la información suficiente para que se recupere la conformación tridimensional, y con ella la función biológica, en el proceso de renaturalización.