|
|
CURSO DE BIOLOGÍAAlejandro Porto Andión |
|
|
|
|
|
|
|
Inicio Temas de Herencia Aula virtual |
|
|
|
|
![]()
TEMA 19: GENÉTICA MOLECULAR.
1.- INTRODUCCIÓN: EL CONCEPTO CLÁSICO DE GEN Y EL NACIMIENTO DE LA GENÉTICA MOLECULAR.
El redescubrimiento de los trabajos de Gregor Mendel en el año 1900 y la amplia generalización de sus conclusiones a la que condujeron los trabajos de genetistas como Morgan, Sturtevant y Muller en las primeras décadas del S. XX, trajeron como consecuencia la aceptación prácticamente universal de los principios mendelianos de la herencia biológica. Esta aceptación propició grandes avances en el conocimiento de los procesos genéticos que afectan tanto a las células individuales como a los organismos pluricelulares y a las poblaciones de seres vivos.
El gran bagaje de
conocimientos acumulados, que globalmente configuran lo que se ha
dado en llamar genética clásica, no sólo ha elevado nuestro
nivel de comprensión de los sistemas vivos hasta cotas insospechadas
pocos años antes, sino que su aplicación en campos como la
agricultura y la medicina ha resultado enormemente beneficiosa para
la humanidad. Sin embargo, durante todo este período, que abarca la
primera mitad del S. XX, el concepto fundamental de la genética, el
gen, permanecía desprovisto de todo contenido material. Aunque la
teoría cromosómica de la herencia había establecido con claridad la
localización de los genes en el núcleo celular y, más concretamente,
en los cromosomas, los genetistas clásicos desconocían por completo
la naturaleza físico-química del gen, así como los mecanismos por
los que éste, desde su sede en el núcleo celular, era capaz de
dirigir la maquinaria bioquímica de la célula y de replicarse con
exactitud a lo largo de muchas generaciones celulares. El gen
mendeliano era una entidad indivisible y abstracta cuya existencia
era reconocida por sus efectos sobre células y organismos aunque su
naturaleza material continuase siendo un misterio. A la pregunta
"¿qué es un gen?"
un genetista clásico probablemente respondiese que el gen es
"algo"
capaz de controlar un carácter hereditario, de replicarse a sí mismo
fielmente en las sucesivas generaciones celulares, de recombinar con
otros genes en el proceso de división celular meiótica y de cambiar
globalmente su estructura para producir una alternativa diferente
del carácter que controla en el proceso conocido como mutación.
Conocer la naturaleza física del gen y los mecanismos moleculares
mediante los cuales éste se replica y controla un carácter
hereditario, aunque de indudable interés intelectual para el
genetista clásico, no es el objetivo de su trabajo, puesto que las
teorías y predicciones experimentales que formula acerca de los
mecanismos de la herencia, y el éxito de las mismas, no dependen de
estos conocimientos.
En los años 40, el panorama hasta aquí dibujado cambió radicalmente cuando un nutrido grupo de científicos, cuya formación y motivaciones eran muy diferentes de las de los genetistas clásicos, comenzó a interesarse por la naturaleza del gen. Se trataba de investigadores que se habían formado en el campo de las ciencias físicas y que estaban escasamente familiarizados con los conocimientos acumulados en las décadas precedentes por los genetistas clásicos e, incluso, con la Biología en general. Por razones que desarrollaremos a continuación, este grupo de científicos centró su interés en la resolución de un único problema: la base física de la información genética.
Los antecedentes de este movimiento intelectual hay que buscarlos en la exposición, por parte de Niels Bohr (Figura 19.1), uno de los más ilustres físicos del S. XX, de la idea de que algunos de los fenómenos biológicos podrían no ser completamente explicables en función de conceptos físicos y químicos convencionales. En opinión de Bohr, y de algunos de sus discípulos, la herencia biológica era claramente uno de estos fenómenos.
Las ideas de Bohr, que en algún momento fueron mal interpretadas y tergiversadas con la intención de resucitar la vieja doctrina filosófica del vitalismo, no llegaron a calar hondo entre la comunidad científica hasta que en 1945 (inmediatamente después del final de la segunda guerra mundial), Erwin Schrödinger (Figura 19.2), uno de los padres de la mecánica cuántica, publicó un pequeño ensayo titulado "¿Qué es la vida?", en el que dichas ideas eran recogidas y desarrolladas de manera mucho más rigurosa. Para Schrödinger, el único problema real, aquel en el que las explicaciones físicas convencionales podrían resultar insuficientes, era la naturaleza física del gen.
 Las estimaciones de los
tamaños de los genes que se deducían de los análisis realizados por
los genetistas clásicos en la mosca del vinagre (Drosophila
melanogáster) indicaban que éstos eran similares a los de las
mayores moléculas conocidas. Si, como apuntaban estas estimaciones,
el gen no era más que un tipo particular de molécula, se trataba, en
opinión de Schrödinger, de una molécula muy especial. En primer
lugar, el gen demostraba ser una molécula altamente estable, capaz
de conservar su estructura específica, y por lo tanto su contenido
informativo, durante largos períodos de tiempo y en un ambiente
químicamente heterogéneo como es el ambiente celular. En segundo
lugar, lo que resultaba todavía más desconcertante, la
"molécula génica"
era capaz de dar lugar a copias fieles de sí misma y transmitirlas
sin alteración a lo largo de innumerables generaciones celulares. No
existía ninguna molécula conocida que reuniera estas
características.
Las estimaciones de los
tamaños de los genes que se deducían de los análisis realizados por
los genetistas clásicos en la mosca del vinagre (Drosophila
melanogáster) indicaban que éstos eran similares a los de las
mayores moléculas conocidas. Si, como apuntaban estas estimaciones,
el gen no era más que un tipo particular de molécula, se trataba, en
opinión de Schrödinger, de una molécula muy especial. En primer
lugar, el gen demostraba ser una molécula altamente estable, capaz
de conservar su estructura específica, y por lo tanto su contenido
informativo, durante largos períodos de tiempo y en un ambiente
químicamente heterogéneo como es el ambiente celular. En segundo
lugar, lo que resultaba todavía más desconcertante, la
"molécula génica"
era capaz de dar lugar a copias fieles de sí misma y transmitirlas
sin alteración a lo largo de innumerables generaciones celulares. No
existía ninguna molécula conocida que reuniera estas
características.
Schrödinger sugería en su ensayo que la molécula génica podría ser un gran cristal aperiódico consistente en la sucesión de unos cuantos elementos isómeros y que la naturaleza exacta de esta sucesión constituiría el código genético. Apuntaba, además, que, por el hecho de que las propiedades exhibidas por la molécula génica no resultaran explicables desde el punto de vista de las leyes físicas conocidas hasta la fecha, no había que presuponer que dicha molécula eludiese dichas leyes. Por el contrario, estas propiedades podrían implicar la existencia de "otras leyes físicas", desconocidas por el momento, que, una vez descubiertas, formarían parte integral de esta ciencia junto con las ya conocidas.
La propuesta de Schrödinger tuvo efectos inmediatos. Los físicos de la época se encontraban sumidos en un gran malestar profesional, probablemente relacionado con el uso bélico que se había hecho de sus investigaciones al final de la segunda guerra mundial, y estaban deseosos de dirigir sus esfuerzos hacia nuevas fronteras del conocimiento. En este contexto, un físico del prestigio de Erwin Schrödinger expone la idea de que el estudio de la materia viva, y más concretamente del gen, podría revelar la existencia de "otras leyes físicas" que aguardaban ahí a que alguien las descubriese. Animados por esta posibilidad, un buen número de físicos decidió abandonar el campo de la investigación para el que habían sido formados y "desembarcaron" en las ciencias biológicas con el objetivo de esclarecer la base físico-química de la información genética.
2.- LA NATURALEZA DEL MATERIAL HEREDITARIO. 
En 1868, un médico alemán llamado Friedich Miescher (Figura 19.3) descubrió, cuando analizaba la composición de núcleos de células del pus procedente de vendajes quirúrgicos, un cuarto tipo de sustancia que se añadía a los ya por entonces conocidos glúcidos, lípidos y proteínas como componente esencial de la materia viva. Se trataba de una sustancia ácida, rica en fósforo, que Miescher denominó nucleína y que poco después empezó a conocerse con el nombre de ácido nucleico, denominación esta que hacía referencia tanto a su carácter ácido como a su localización en el núcleo celular.
Resulta paradójico que esta sustancia fuese descubierta sólo tres años después de que Mendel estableciese el concepto de gen y que hubiesen de transcurrir otros noventa hasta que dicha sustancia fuese reconocida universalmente como su soporte material.
El conocimiento preciso de la química elemental de los ácidos nucleicos se demoró bastante con respecto al de otras biomoléculas orgánicas, probablemente debido a su mayor complejidad estructural. En los primeros años del S. XX fueron identificados sus componentes moleculares (pentosas, bases nitrogenadas y ácido fosfórico). En la década de los años 20 se descubrió cómo se unen entre sí estos componentes para dar lugar a los nucleótidos y cómo se enlazan éstos para dar lugar a los ácidos nucleicos. Por la misma época se pudo reconocer la existencia de dos tipos principales de ácido nucleico (DNA y RNA) así como las diferencias estructurales entre ellos. Todavía a comienzos de los años 40 se desconocía el hecho de que los ácidos nucleicos eran en realidad macromoléculas que consistían en largas cadenas polinucleotídicas, formadas por centenares, miles e incluso millones de nucleótidos unidos por puentes fosfodiéster. Cuando en 1945 Schrödinger propone a sus colegas físicos investigar la naturaleza del material genético, era comúnmente aceptado que la molécula de DNA consistía en un tetranucleótido cíclico, formado por los cuatro desoxirribonucleótidos conocidos. La idea del tetranucleótido provenía de los análisis cuantitativos realizados sobre DNAs de unas pocas especies de seres vivos, que indicaban que en todos ellos las 4 bases nitrogenadas (A, T, G y C) se encontraban en cantidades iguales.
Por otra parte, en la teoría cromosómica de la herencia se establecía ya la localización de los genes a lo largo de los cromosomas, dentro del núcleo celular. El análisis químico de los cromosomas indicaba que éstos estaban compuestos por DNA y proteínas a partes aproximadamente iguales. Todo indicaba pues que alguna de estas dos sustancias debía ser el material hereditario.
La química elemental de las proteínas era bien conocida ya cuando empezó a plantearse el problema de la naturaleza del gen. Aunque sólo se habían dado los primeros pasos en la determinación de su estructura tridimensional, se había establecido ya con claridad que las moléculas proteicas consistían en largas cadenas de aminoácidos unidos mediante enlaces peptídicos. También se había puesto de manifiesto que la identidad química de cada molécula proteica vendría dada por la naturaleza y posición de los diferentes restos de aminoácidos a lo largo de la cadena polipeptídica, es decir, por su estructura primaria.
En este contexto histórico, con un conocimiento bastante avanzado de la estructura química de las proteínas y relativamente pobre de la de los ácido nucleicos, un buen número de investigadores se decantó inicialmente por las proteínas como principales candidatas a constituir la base química de la herencia. Resultaba claro para ellos que las largas cadenas de aminoácidos de las proteínas respondían mejor a la idea del gran cristal aperiódico formulada por Schrödinger que el pequeño tetranucleótido cíclico en que parecía consistir la molécula de DNA. Según su punto de vista, la información genética podría estar cifrada en forma de diferentes secuencias de aminoácidos, siendo éstos los "elementos isómeros" a los que Schrödinger hacía referencia. En los próximos apartados de este capítulo comprobaremos que el tiempo y los hechos vinieron a quitarles la razón.
2.1.- EL EXPERIMENTO DE AVERY.
 La primera prueba de que
el DNA era después de todo el material genético, fue obtenida en
1944 por Oswald T. Avery, y sus colaboradores C.M. McLeod y M.J.
McCarty en el transcurso de un trabajo experimental en el que
trataban de encontrar explicación a un fenómeno observado algunos
años antes por el médico británico F. Griffith (Figura
19.4).
La primera prueba de que
el DNA era después de todo el material genético, fue obtenida en
1944 por Oswald T. Avery, y sus colaboradores C.M. McLeod y M.J.
McCarty en el transcurso de un trabajo experimental en el que
trataban de encontrar explicación a un fenómeno observado algunos
años antes por el médico británico F. Griffith (Figura
19.4).
En 1928 F. Griffith estudiaba el proceso de infección en ratones por Streptococcus pneumoniae, más conocido como "neumococo", una bacteria que se encuentra entre los agentes causantes de la neumonía humana y que resulta especialmente patógena para el ratón: la inyección en un ratón de esputos procedentes de un paciente afectado de neumonía neumocócica le ocasiona a aquél la muerte en menos de 24 horas. El neumococo debe su carácter patógeno a una cápsula de polisacáridos que lo protege de los mecanismos de defensa del animal infectado. Griffith había aislado una cepa mutante de esta especie, que había perdido su capacidad para sintetizar esta cápsula y que resultaban por lo tanto vulnerables a dichos mecanismos de defensa: los ratones inoculados con bacterias de esta cepa no contraían la neumonía y por consiguiente sobrevivían. Ambas variantes podían distinguirse una de la otra con facilidad debido al aspecto de las colonias que formaban en las placas de cultivo, que tenían aspecto brillante (S) en la variante patógena común y aspecto rugoso (R) en la variante mutante no patógena. El aspecto brillante o rugoso de las colonias era también una consecuencia de la presencia o ausencia respectivamente de la mencionada cápsula de polisacáridos.
En el curso de sus investigaciones Griffith descubrió con sorpresa que los ratones inoculados con mutantes R no patógenos mezclados con una muestra de bacterias S patógenas previamente muertas por efecto del calor, contraían la neumonía y morían a las pocas horas. Las bacterias recuperadas de la sangre de los ratones muertos habían recuperado su capacidad para sintetizar la cápsula de polisacáridos y con ello su carácter patógeno y el aspecto brillante de las colonias a las que daban lugar. El contacto con las bacterias S había producido en las bacterias R una transformación RàS que se transmitía a las sucesivas generaciones celulares. Años más tarde Griffith comprobó que no era imprescindible que el contacto entre las dos cepas bacterianas se produjese en el interior del ratón: la transformación también se producía en cultivos de bacterias R que crecían en contacto con bacterias S muertas. Más significativo aún resultó el hecho de que la transformación se produjese como consecuencia del contacto de cultivos de bacterias R creciendo en contacto con un "extracto libre de células" de bacterias S, es decir, no era imprescindible la estructura celular intacta de las bacterias S muertas sino que una disolución de sus componentes moleculares solubles era suficiente.

Los experimentos de Griffith fueron el punto de partida del trabajo de Avery, McLeod y McCarthy, que se plantearon identificar, en el extracto libre de células que se ha mencionado, la naturaleza química del "principio transformante" responsable del fenómeno observado. Para ello llevaron a cabo un fraccionamiento sistemático del extracto libre de células y ensayaron la capacidad transformante de las distintas fracciones sobre cultivos de bacterias R. Tras ensayar con distintas fracciones del extracto (lípidos, glúcidos, proteínas, etc.) con resultados negativos, comprobaron que la fracción que contenía los ácidos nucleicos inducía eficazmente la transformación. Un fraccionamiento ulterior llevó a la conclusión de que el principio transformante buscado no era otro que el DNA bacteriano: pequeñísimas cantidades de este DNA purificado eran suficientes para transformar las bacterias R en bacterias S. Avery y sus colaboradores demostraron también que el DNA de las bacterias transformadas y de sus descendientes podía a su vez inducir la transformación en otras bacterias R y que en sucesivos ciclos de transformación como los descritos se mantenía esta capacidad.
Tras estas experiencias las
conclusiones de Avery (Figura
19.6) estaban cada vez más claras: el DNA de las bacterias S muertas
era la sustancia que contenía la información necesaria para hacer que
las bacterias R y su descendencia recuperasen su capacidad para
sintetizar su cápsula de polisacáridos y con ella su carácter patógeno,
es decir, el DNA era el material genético de Streptococcus pneumoniae.
La publicación de los resultados de Avery, McLeod y McCarthy en 1944 provocó una oleada de escepticismo crítico entre la comunidad científica de la época. Como ya se ha dicho, la mayoría de los investigadores apuntaba a las proteínas como principales candidatas a constituir la base química del gen. Las principales objeciones incidían en el hecho de que las técnicas al uso de fraccionamiento y purificación de macromoléculas no eran eficaces al 100%, de manera que una pequeña cantidad de proteínas contaminando el extracto de DNA purificado pudiera ser la responsable de la actividad transformante de éste. Analizada desde la perspectiva actual esta objeción parece claramente formulada ad hoc en defensa de una idea previa demasiado firmemente asentada: “tenían que ser las proteínas”. En efecto, si el material genético consiste en proteínas, no parece lógico afirmar que la actividad transformante reside en una mínima cantidad de proteína contaminante en la fracción de DNA purificado y no en la que contiene la mayor parte de las proteínas de la célula. Sin embargo, tales críticas propiciaron la realización de nuevos controles experimentales que siempre confirmaron las conclusiones precedentes. El equipo de Avery trató el extracto el DNA purificado de las bacterias S con proteasas (enzimas que degradan las proteínas) sin que esto afectara a su actividad transformante. Por otra parte, el tratamiento con desoxirribonucleasas (enzimas que degradan el DNA) destruía en pocos minutos cualquier rastro de dicha actividad. Otros experimentos, realizados por R.D. Hotchkiss, confirmaron que la actividad transformante del DNA no se restringía al carácter virulento o no de las cepas bacterianas, ni al aspecto liso o rugoso de sus colonias, sino que también operaba de manera análoga para otros caracteres hereditarios, como la resistencia a distintos tipos de antibióticos.
Los nuevos resultados experimentales no consiguieron diluir el escepticismo reinante. Si nueve años después de la publicación de los trabajos de Avery la comunidad científica aceptó por fin el papel del DNA como molécula portadora de la información genética, no fue por que se hubiesen realizado nuevos controles con resultados más convincentes, sino porque los grandes avances que se produjeron en este período acerca de la química de los ácidos nucleicos disiparon cualquier duda al respecto.
2.2.- LA REGLA DE EQUIVALENCIA DE CHARGAFF.
La principal dificultad a la hora de reconocer al DNA un papel relevante en la herencia biológica residía en que nadie comprendía, a la luz de los conocimientos existentes acerca de la química de los ácidos nucleicos, cómo podía desempeñarlo. Ya se ha comentado que a comienzos de los años cuarenta se consideraba que la molécula de DNA era un tetranucleótido cíclico formado por los cuatro desoxirribonucleótidos posibles (dAMP, dGMP, dTMP y dCMP). Incluso cuando en el curso de esta década se estableció que el DNA era en realidad una macromolécula con peso molecular muy superior al que cabría esperar de un simple tetranucleótido, se interpretó que éste vendría a ser la unidad monomérica repetitiva que formaba los grandes polímeros de DNA, de manera similar a lo que representaba la glucosa en los polímeros monótonos de almidón y glucógeno. Era evidente que una macromolécula con estas características no podía ser el cristal aperiódico de Schrödinger.
 La persistencia de
la idea del tetranucleótido tenía su razón de ser en que los análisis de
la composición química de DNA procedente de diferentes fuentes
biológicas parecían arrojar siempre proporciones equimolares de las
cuatro bases nitrogenadas (A, G, T y C). A finales de los años 40 Erwin
Chargaff (Figura
19.7) adaptó las recién descubiertas técnicas de cromatografía sobre
papel a la separación y cuantificación de los componentes de los ácidos
nucleicos y las empleó en el análisis de diferentes muestras de DNA. La
mayor precisión de estas técnicas le permitió comprobar que las cuatro
bases nitrogenadas no se encontraban necesariamente en proporciones
exactamente iguales. Además, Chargaff encontró que la composición en
bases nitrogenadas difiere ampliamente según la procedencia biológica de
la muestra de DNA.
La persistencia de
la idea del tetranucleótido tenía su razón de ser en que los análisis de
la composición química de DNA procedente de diferentes fuentes
biológicas parecían arrojar siempre proporciones equimolares de las
cuatro bases nitrogenadas (A, G, T y C). A finales de los años 40 Erwin
Chargaff (Figura
19.7) adaptó las recién descubiertas técnicas de cromatografía sobre
papel a la separación y cuantificación de los componentes de los ácidos
nucleicos y las empleó en el análisis de diferentes muestras de DNA. La
mayor precisión de estas técnicas le permitió comprobar que las cuatro
bases nitrogenadas no se encontraban necesariamente en proporciones
exactamente iguales. Además, Chargaff encontró que la composición en
bases nitrogenadas difiere ampliamente según la procedencia biológica de
la muestra de DNA.
Puede resultar hoy extraño el que no se detectase antes esta amplia variación. Lo cierto es que en las muestras de DNA analizadas hasta entonces, la mayoría de ellas procedentes de organismos eucariontes, las proporciones de las distintas bases nitrogenadas oscilaban unos pocos puntos porcentuales con respecto al 25% que exigía la hipótesis del tetranucleótido, de manera que los primitivos métodos de análisis no permitían distinguir los resultados de los que se obtendrían si efectivamente las cuatro bases se encontrasen en proporciones equimolares. En los años subsiguientes se analizaron muestras de DNA procedentes de diferentes especies bacterianas, pudiendo comprobarse que en las células procariotas el espectro de variación de la composición en bases nitrogenadas es todavía mucho más amplio que en las células eucariotas.
La publicación de los resultados de Chargaff no sólo trajo consigo el rechazo definitivo de la hipótesis del tetranucleótido, sino que provocó un vuelco en la opinión de los científicos acerca del papel de los ácidos nucleicos. En efecto, si el DNA no era después de todo un polímero monótono, se había esfumado el principal inconveniente de la candidatura de esta macromolécula a constituir la base físico-química de la herencia biológica. Un largo polímero formado por cuatro tipos de nucleótidos en diferentes ordenaciones sí podía ser el cristal aperiódico formado por unos pocos elementos isómeros que Schrödinger había sugerido. Además, el hecho de que la composición en bases nitrogenadas del DNA variase ampliamente de unas especies a otras podía ser un reflejo de su especificidad biológica. La información genética podría muy bien estar cifrada en forma de la secuencia específica de bases nitrogenadas de la cadena polinucleotídica y el fenómeno de la mutación podría explicarse como un cambio fortuito en dicha secuencia.
Los analistas de la historia de la Biología molecular dudan a la hora de atribuir a un científico en particular la formulación de las ideas precedentes. Más bien se inclinan por afirmar que, a partir de 1950, la teoría parecía “flotar en el ambiente”, provocando una gran efervescencia investigadora alrededor de la molécula de DNA en los años sucesivos.
Además de todo lo dicho hasta ahora, los resultados publicados por Chargaff contenían una información adicional acerca de la molécula de DNA, que andando el tiempo resultó de importancia capital: a pesar de la amplia variación encontrada entre las muestras de DNA de diferentes especies, en todas ellas la proporción molar entre el total de bases púricas y el de bases pirimídicas era próxima a 1. Lo mismo parecía ocurrir con la adenina y la timina por una parte y la guanina y la citosina por otra. Esta afirmación, conocida como la “regla de equivalencia de Chargaff”, revela un rasgo esencial de la molécula de DNA: aunque la composición en bases nitrogenadas de este polímero puede variar sin ninguna restricción, se cumple siempre que el número de adeninas es igual al de timinas y el de guaninas igual al de citosinas; y, como corolario, que el número de bases púricas es igual al de bases pirimídicas.
[A] = [T]
[G] = [C]
-----------------------------------
[A+G] = [T+C]
Aunque cuando publicó sus resultados Chargaff manifestó que la constancia de estas proporciones era algo “digno de resaltar” (“noticeable”), se mostró extraordinariamente prudente a la hora de considerar si se trataba de una simple coincidencia o si realmente revelaba algún rasgo importante de la molécula de DNA. Realmente, hubiera resultado asombroso que Chargaff pudiera intuir el significado de su regla de equivalencia con la información de que disponía en 1950.
2.3.- EL EXPERIMENTO DE HERSHEY Y CHASE.
Entre los microorganismos cuyo estudio contribuyó en mayor medida a la comprensión de la estructura y función del gen hay que destacar a los virus, que serán analizados en detalle en un capítulo posterior. A comienzos de los años 50 del S. XX los virus eran ya reconocidos como parásitos intracelulares obligados, con un grado de organización inferior al celular, que se reproducían dentro de las células a las que parasitaban. Se les relacionaba acertadamente con un buen número de enfermedades humanas de las que eran responsables y se había detectado su presencia en distintas especies animales y vegetales. También se habían descubierto virus que parasitaban y destruían células bacterianas, los llamados bacteriófagos.
La estructura de los virus es extremadamente simple en comparación con la de los organismos con organización celular: los más simples constan de una cápside de naturaleza proteica en la que se encierra un ácido nucleico que puede ser DNA o RNA.
 En 1952 Alfred D. Hershey y Marta Chase
(Figura
19.8) diseñaron un experimento con el objeto de elucidar los
detalles del proceso de infección de células bacterianas de la especie
Escherichia Coli por bacteriófagos T4 (Figura 19.8b). Las partículas infecciosas
de este fago están compuestas exclusivamente por DNA y proteínas. Hershey y Chase querían saber como se comportaba uno y otro tipo de
macromoléculas durante el proceso de infección. Para ello, idearon una
ingeniosa técnica de marcaje mediante isótopos radiactivos. Se
percataron de que en las partículas virales la práctica totalidad de los
átomos de fósforo se encontraban en el DNA (en los grupos fosfato de la
cadena polinucleotídica) mientras que la práctica totalidad de los
átomos de azufre se encontraban en las proteínas (en los aminoácidos
metionina y cisteína). Así, decidieron utilizar los isótopos radiactivos
32P y 35S para delatar respectivamente la
presencia de DNA y de proteínas. Para obtener partículas víricas
marcadas permitieron el crecimiento de un cultivo de fagos T4 sobre
células de E. coli en un medio en el que la única fuente de fósforo
eran iones fosfato (PO43-) marcado con 32P,
de manera que este isótopo se incorporaba a todas las biomoléculas de
las células bacterianas y de los fagos que se reproducían en su
interior. Por otra parte, hicieron lo propio con fagos obtenidos en un
cultivo con iones sulfato (SO42-) marcado con
35S, que se incorporaría igualmente a las biomoléculas de
bacterias y fagos. De este modo, una vez aislados y purificados los
fagos obtenidos en uno y otro cultivo, dispusieron de dos cepas virales,
una de ellas con el DNA marcado con 32P y otra con las
proteínas marcadas con 35S.
En 1952 Alfred D. Hershey y Marta Chase
(Figura
19.8) diseñaron un experimento con el objeto de elucidar los
detalles del proceso de infección de células bacterianas de la especie
Escherichia Coli por bacteriófagos T4 (Figura 19.8b). Las partículas infecciosas
de este fago están compuestas exclusivamente por DNA y proteínas. Hershey y Chase querían saber como se comportaba uno y otro tipo de
macromoléculas durante el proceso de infección. Para ello, idearon una
ingeniosa técnica de marcaje mediante isótopos radiactivos. Se
percataron de que en las partículas virales la práctica totalidad de los
átomos de fósforo se encontraban en el DNA (en los grupos fosfato de la
cadena polinucleotídica) mientras que la práctica totalidad de los
átomos de azufre se encontraban en las proteínas (en los aminoácidos
metionina y cisteína). Así, decidieron utilizar los isótopos radiactivos
32P y 35S para delatar respectivamente la
presencia de DNA y de proteínas. Para obtener partículas víricas
marcadas permitieron el crecimiento de un cultivo de fagos T4 sobre
células de E. coli en un medio en el que la única fuente de fósforo
eran iones fosfato (PO43-) marcado con 32P,
de manera que este isótopo se incorporaba a todas las biomoléculas de
las células bacterianas y de los fagos que se reproducían en su
interior. Por otra parte, hicieron lo propio con fagos obtenidos en un
cultivo con iones sulfato (SO42-) marcado con
35S, que se incorporaría igualmente a las biomoléculas de
bacterias y fagos. De este modo, una vez aislados y purificados los
fagos obtenidos en uno y otro cultivo, dispusieron de dos cepas virales,
una de ellas con el DNA marcado con 32P y otra con las
proteínas marcadas con 35S.
Con las cepas virales obtenidas Hershey y Chase procedieron a infectar dos cultivos de E. coli no marcados radiactivamente (cada uno de ellos con una cepa diferente). Tras permitir la infección por un corto período de tiempo separaron por centrifugación las partículas víricas que no habían conseguido adherirse a ninguna célula y volvieron a suspender las bacterias infectadas en un medio de cultivo nuevo. A continuación sometieron esta suspensión de bacterias infectadas a una violenta agitación por medio de un agitador Waring (un dispositivo que genera fuertes turbulencias en el líquido sobre el que actúa), de manera que las partículas víricas se desprendían (eran literalmente arrancadas) de la superficie celular sobre la que se habían fijado. Se procedió entonces a centrifugar la suspensión con el objeto de separar las bacterias, que se depositaban en el fondo del tubo de la centrífuga, de las partículas víricas sueltas, que permanecían en el líquido sobrenadante. Seguidamente, se midió en un contador Geiger la fracción total de radiactividad que se había depositado en el fondo del tubo y la que permanecía en el líquido sobrenadante. Por último, se ensayó la capacidad de las bacterias infectadas para producir nuevos fagos descendientes en su interior. Los resultados obtenidos fueron los siguientes:
-
En el cultivo bacteriano infectado con fagos marcados con 32P, es decir, con DNA marcado radiactivamente, la mayor parte de la radiactividad se había depositado en el fondo del tubo de la centrífuga. La fracción que no lo había hecho así se encontraba en el líquido sobrenadante de la primera centrifugación, es decir, que se encontraba en las partículas víricas que no habían conseguido infectar a ninguna célula.
-
En el cultivo bacteriano infectado con fagos marcados con 35S, es decir, con proteínas marcadas radiactivamente, la mayor parte de la radiactividad permanecía en el líquido sobrenadante obtenido tras la agitación violenta. La fracción restante correspondía a estructuras de la cápside viral que no se habían desprendido de la superficie celular por estar demasiado intensamente ligadas a ella.
-
En ambos cultivos las bacterias infectadas recuperadas tras la agitación violenta conservaban prácticamente intacta su capacidad para dar lugar a nuevas progenies virales a su vez con capacidad infectiva.
Hershey y Chase extrajeron rápidamente conclusiones de estos resultados (Figura 19.8b. Hacer click para ver aumentado). De los dos componentes de la partícula vírica sólo el DNA penetraba en el interior de la célula durante el proceso de infección (por eso el 32P aparecía asociado a la fracción celular tras la centrifugación). Las proteínas del fago permanecían en el exterior de la célula durante todo el proceso de infección y se desprendían de la superficie celular por agitación (por eso el 35S aparecía en el líquido sobrenadante). Es decir, la partícula vírica infecciosa se fija a la superficie celular y de alguna manera “inyecta” su DNA en el interior de la célula. La cápside proteica, una vez inyectado en la bacteria el DNA que albergaba en su interior, ya no resulta más necesaria en el proceso de infección, como prueba el hecho de que la eliminación de estos “fagos fantasma” por agitación no altere la capacidad de las células infectadas para dar lugar a nuevas progenies virales. Es la molécula de DNA vírico la que, una vez dentro de la célula, parece tomar el control de su metabolismo para que éste se ponga al servicio del virus y comience a fabricar nuevas partículas infecciosas atendiendo a las instrucciones cifradas en esa misma molécula. En otras palabras: el DNA es el material genético del bacteriófago T4.
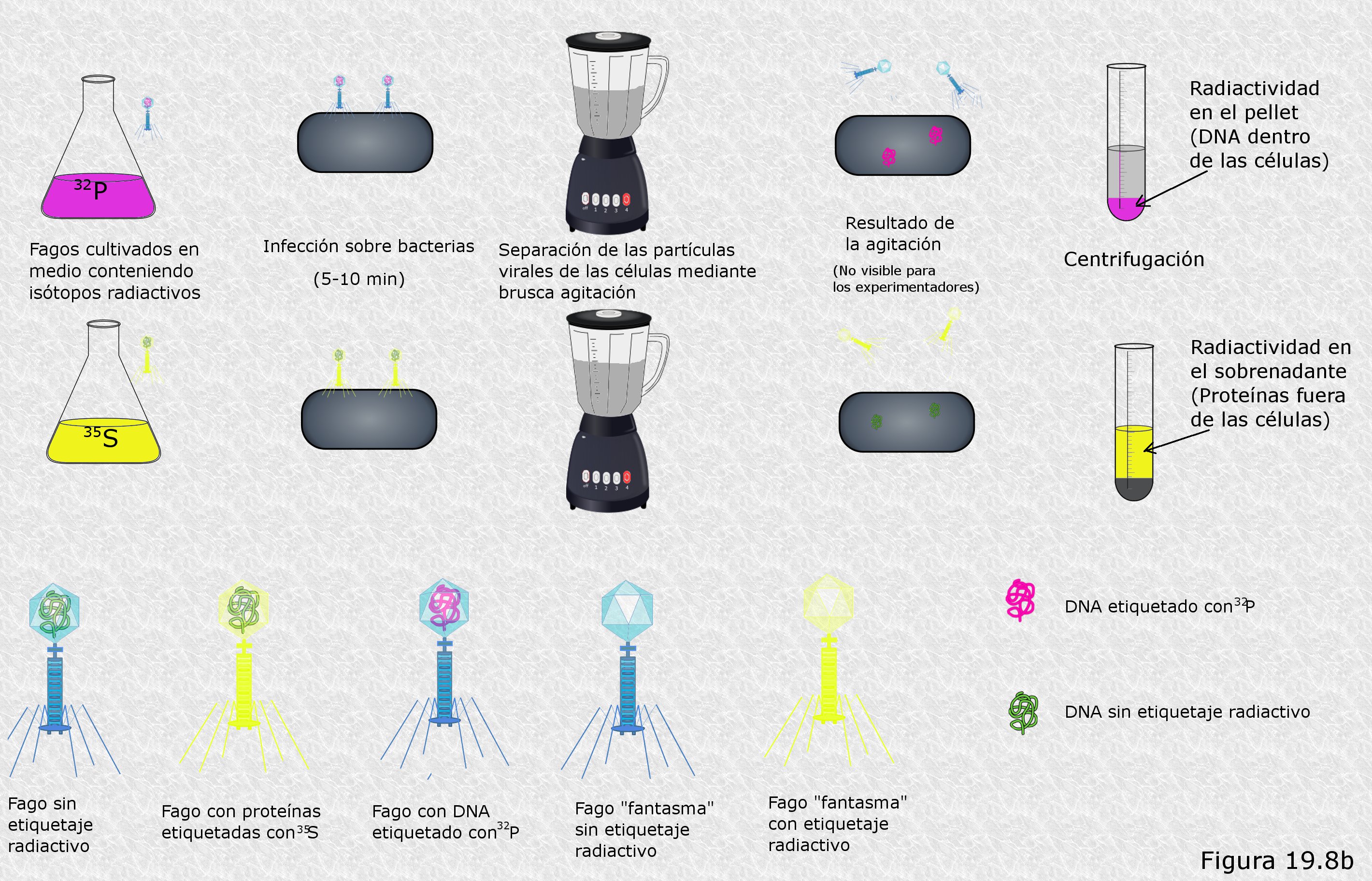
Los resultados obtenidos por Hershey y Chase eran especialmente concluyentes en lo que se refiere al papel del DNA como molécula de la herencia, pues en su experimento queda claro que el único nexo material entre dos generaciones sucesivas de bacteriófagos es una simple molécula de DNA.
Por otra parte, las conclusiones de este experimento concordaban y reforzaban las obtenidas ocho años antes por el equipo de Avery. Pero ahora el escenario había cambiado: los recientes descubrimientos acerca de la química de los ácidos nucleicos junto con los resultados obtenidos por Chargaff sobre la composición en bases nitrogenadas de diferentes muestras de DNA habían preparado el terreno para que estas conclusiones gozaran de una aceptación mucho mayor que la que se deparó a las publicadas por Avery. Probablemente, la publicación del trabajo de Hershey y Chase en el otoño de 1952 sirvió de estímulo para que un buen número de investigadores se concentraran durante los meses siguientes en elucidar la estructura tridimensional de la molécula de DNA.
2.4.- LA ESTRUCTURA DEL DNA: LA DOBLE HÉLICE.

 Una de las técnicas de análisis que
resultó de mayor utilidad para la comprensión de la estructura
tridimensional de las biomoléculas fue la cristalografía de difracción
de rayos X. Como ya se ha comentado en un capítulo anterior, esta
técnica fue aplicada con éxito al estudio de la conformación
tridimensional de las proteínas.
Una de las técnicas de análisis que
resultó de mayor utilidad para la comprensión de la estructura
tridimensional de las biomoléculas fue la cristalografía de difracción
de rayos X. Como ya se ha comentado en un capítulo anterior, esta
técnica fue aplicada con éxito al estudio de la conformación
tridimensional de las proteínas.
El primer investigador en dirigir su atención a la estructura tridimensional del DNA fue William Astbury (pionero también en la aplicación de la cristalografía de RX al análisis de la estructura de las proteínas). Ya en 1945, tras deducir de la elevada densidad de las muestras de DNA que los nucleótidos debían encontrarse en la cadena polinucleotídica fuertemente empaquetados o “apilados” unos sobre otros, obtuvo algunos difractogramas de RX de la molécula de DNA que, a pesar de su baja calidad, demostraban que efectivamente los nucleótidos se encontraban apilados con una separación de 0,34 nm entre cada dos restos sucesivos.
A comienzos de los años 50 tres centros de investigación rivalizaban en el análisis de la estructura tridimensional de las biomoléculas mediante cristalografía de RX. Uno de ellos era el Instituto de Tecnología de California (Cal Tech), cuya división de química, dirigida por Linus Pauling (Figura 19.10), se había apuntado varios éxitos notables en el descubrimiento de la estructura secundaria de las proteínas. Otro era el Laboratorio Cavendish de la Universidad de Cambridge (Inglaterra), dirigido por Sir Willian Lawrence Bragg (Figura 19.9), en el que grandes cristalógrafos como John Kendrew y Max Perutz centraban su atención en el estudio de las estructuras terciaria y cuaternaria de las proteínas globulares. Fue en el Laboratorio Cavendish donde coincidieron a comienzos de 1951 dos jóvenes investigadores, James D. Watson y Francis H.C. Crick, que estaban llamados a ser quienes desvelaran finalmente el misterio del gen. Un tercer grupo se había formado en el King’s College de Londres bajo la jefatura de John Randall, que contaba con la colaboración de Maurice Wilkins y de la experta cristalógrafa Rosalind Franklin (Figura 19.11).

Entre 1951 y 1953 se desató entre estos grupos una especie de carrera por identificar la estructura tridimensional del DNA, carrera que se desbocó por completo cuando la publicación del experimento de Hershey y Chase a finales de 1952 puso a todos los grupos sobre la pista correcta de cual era en realidad la molécula de la herencia. Linus Pauling, en principio favorito para ganar esta carrera dado su enorme prestigio, no tuvo éxito en esta ocasión; publicó, a comienzos de 1953, una propuesta de estructura para el DNA que contenía errores de bulto que obligaron a descartarla inmediatamente después de su publicación. En el King’s College, Rosalind Franklin había desarrollado una técnica que le permitía obtener fibras de DNA altamente orientadas y obtener así difractogramas de RX de una calidad y lujo de detalles muy superiores a los conocidos hasta entonces. Wilkins y Franklin se encontraban a comienzos de 1953 intentando encajar sus datos de RX dentro de un modelo plausible para el DNA. Mientras tanto Watson y Crick trataban de construir su propio modelo tridimensional basándose en difractogramas de una calidad muy inferior. No obstante, su trabajo se encontraba muy avanzado; habían analizado cuidadosamente la estructura de los nucleótidos individuales y se habían percatado de que los datos obtenidos por Chargaff tres años antes, sobre las proporciones de las bases nitrogenadas, debían tener algún significado relevante, lo que probablemente fue una de las claves de su éxito posterior. Fue entonces cuando Jim Watson, durante una conversación con Maurice Wilkins, pudo ver algunos de los difractogramas obtenidos por Rosalind Franklin; la simple inspección visual de aquellos difractogramas proporcionó a Watson las claves que le faltaban para resolver finalmente la estructura del DNA. En pocas semanas Watson y Crick terminaron de encajar sus propios datos con lo que se apreciaba en los difractogramas de Franklin y elaboraron un modelo definitivo que fue publicado en el número de abril de la revista Nature. La carrera había terminado.
 El modelo propuesto por Watson y Crick,
mundialmente conocido como la doble hélice (Figura
19.12), presentaba las siguientes características:
El modelo propuesto por Watson y Crick,
mundialmente conocido como la doble hélice (Figura
19.12), presentaba las siguientes características:
-
La molécula de DNA está formada por dos cadenas polinucleotídicas antiparalelas, es decir, si una cadena se recorre en dirección 5’—›3’, su vecina discurriría en dirección 3’—›5’.
-
Ambas cadenas se encuentran formando un arrollamiento helicoidal de tipo plectonémico, es decir, para separarlas habría que desenrollarlas girando una sobre la otra. El arrollamiento es además dextrógiro.
-
El conjunto forma una estructura cilíndrica con un diámetro constante de 2 nm.
-
Los esqueletos azúcar-fosfato de las cadenas polinucleotídicas se encuentran en el exterior de la estructura, formando lo que serían las guías de una especie de escalera de caracol.
-
Las bases nitrogenadas se proyectan desde los esqueletos azúcar-fosfato hacia el interior de la estructura y se disponen apiladas por pares formando lo que equivaldría a los peldaños de la escalera.
-
Los pares de bases nitrogenadas están formados invariablemente por una purina y una pirimidina. Además, siempre se encuentran enfrentadas adenina con timina por una parte y guanina con citosina por otra.
-
Las dos cadenas polinucleotídicas se encuentran unidas por puentes de hidrógeno entre grupos funcionales de las bases nitrogenadas que forman cada par. Cada adenina forma dos puentes de hidrógeno con la correspondiente timina y cada guanina tres con la citosina. Pares de bases diferentes a los establecidos no podrían formar puentes de hidrógeno.
-
La distancia entre cada par de bases sucesivo es de 0,34 nm. Cada vuelta completa de la hélice (paso de rosca) alberga exactamente 10 pares de nucleótidos, lo que se corresponde con una longitud de 3,4 nm. Ambas periodicidades aparecían reflejadas en los difractogramas.
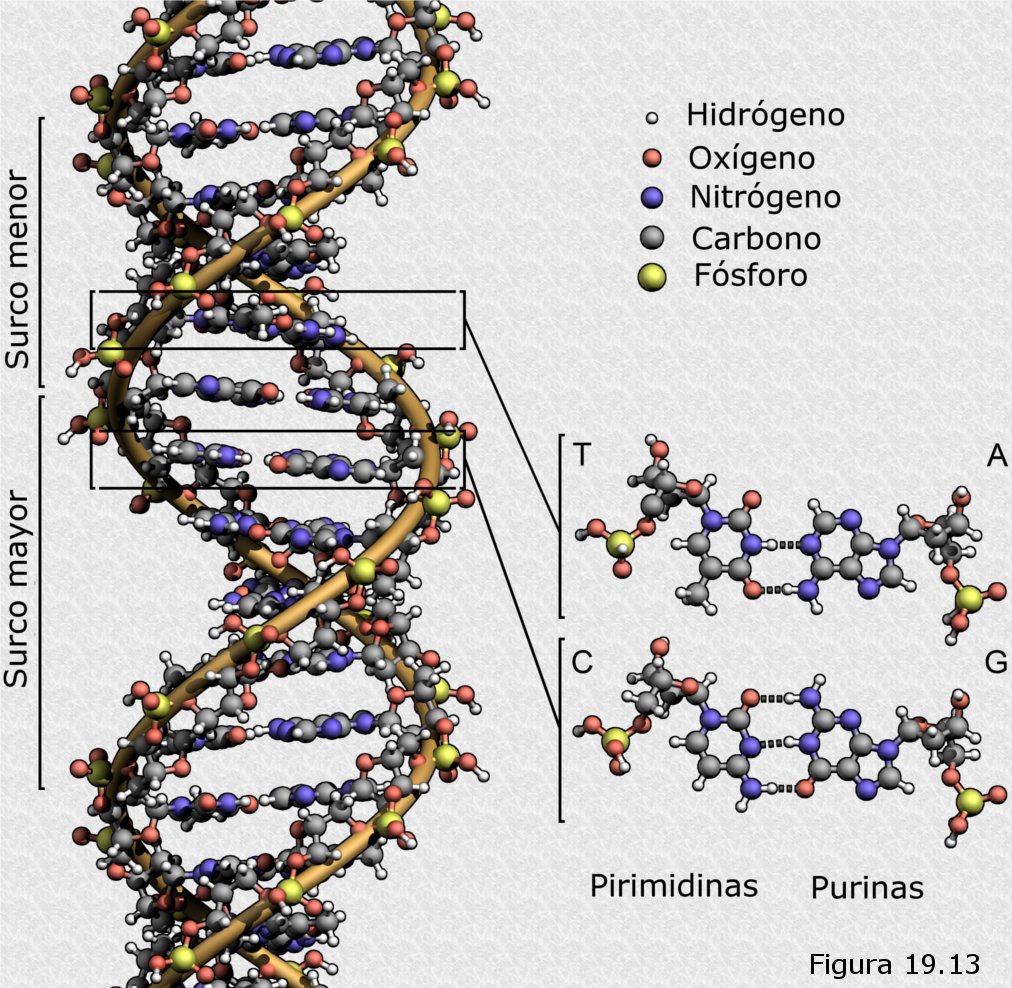
Uno de los aspectos más interesantes del modelo de Watson y Crick residía en que no sólo encajaba con los datos de difracción de RX sino que además proporcionaba una explicación para la hasta entonces desconcertante regla de Chargaff. En efecto, si todos los pares de bases eran necesariamente A-T o G-C, en cualquier muestra de DNA el número de restos de adenina debía ser igual al de timina y el de guanina al de citosina, de lo que se deduce que el número total de bases púricas debería ser igual al de bases pirimídicas. Además, este emparejamiento específico de las bases nitrogenadas podría encerrar un profundo significado biológico, pues, como Watson y Crick sugerían en su artículo en Nature, tal emparejamiento podría ser la base del mecanismo por el que el material genético creaba copias de si mismo en cada ciclo de reproducción celular. La complementariedad interna de la doble hélice, regida por la regla de Chargaff, hacía que cada una de las dos cadenas polinucleotídicas que la formaban pudiera ser utilizada como molde para sintetizar otra con una secuencia de bases complementaria.
La publicación del modelo de Watson y Crick en abril de 1953 y la gran difusión que tuvo en los meses posteriores diluyó rápidamente cualquier resto de escepticismo acerca del papel del DNA como material hereditario, que ya no volvió a ser discutido. Todo ello supuso una auténtica revolución en el seno de las ciencias biológicas y el nacimiento de lo que se dio en llamar biología molecular, área del conocimiento que tuvo un gran desarrollo en las décadas siguientes y que ha contribuido de forma decisiva a nuestra comprensión actual del funcionamiento de los sistemas vivos. Hay que destacar, sin embargo, que los físicos que habían desembarcado en la biología con la aspiración romántica de encontrar “otras leyes físicas” todavía desconocidas se quedaron sin su recompensa. Por el contrario, todo lo que hoy sabemos acerca de cómo los genes se replican y controlan los procesos celulares es perfectamente explicable en términos de procesos físico-químicos convencionales que ya eran conocidos a mediados del S XX. El misterio del gen, que Schrödinger proponía desvelar, no consistía más que en la simple formación y ruptura de puentes de hidrógeno entre las bases nitrogenadas del DNA.
2.5.- ESTRUCTURAS ALTERNATIVAS DEL DNA.
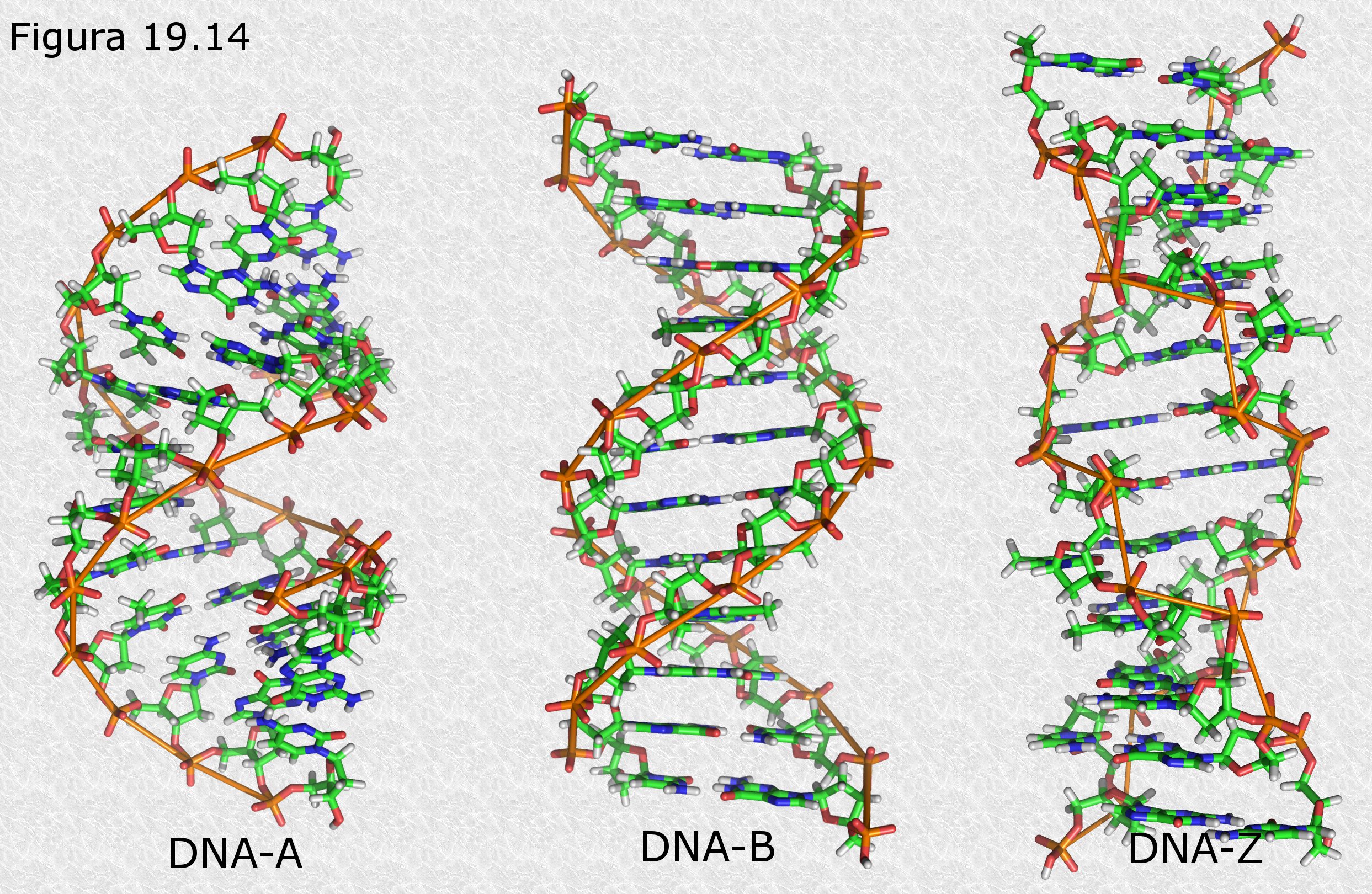
La doble hélice del DNA tal como fue descrita por Watson y Crick representa la estructura más común de esta macromolécula (la llamada forma B). Años más tarde se demostró que el DNA puede existir en al menos dos formas alternativas (la forma A y la forma Z) que difieren ligeramente de la estructura original en aspectos como las distancias entre nucleótidos sucesivos o los ángulos de enlace entre los componentes de estos nucleótidos (Figura 19.14). Sin embargo, se ha comprobado que en estas formas alternativas están presentes los rasgos esenciales del modelo de Watson y Crick, es decir, la estructura helicoidal y el emparejamiento específico de bases.
Por otra parte, se ha podido comprobar que en algunos virus el DNA aparece en estado monocatenario (una sola cadena polinucleotídica por molécula en lugar de dos), lo que constituye una excepción a la primitiva afirmación de que el DNA es siempre una doble hélice de cadenas polinucleotídicas. Sin embargo, se trata de una “excepción que confirma la regla”, ya que incluso en estos virus el DNA pasa por un estado bicatenario transitorio, que resulta imprescindible para su replicación durante el ciclo de reproducción viral.
2.6.- DESNATURALIZACIÓN E HIBRIDACIÓN DEL DNA.
La molécula de DNA es muy estable, gracias a la gran cantidad de puentes de hidrógeno que se establecen entre las bases nitrogenadas a lo largo de las cadenas polinucleotídicas y a las interacciones hidrofóbicas generadas entre los anillos aromáticos apilados de estas bases. Sin embargo, de manera similar a lo que ocurre con las proteínas, la molécula puede desestabilizarse y abandonar su conformación tridimensional característica en doble hélice como respuesta a cambios en el pH o a aumentos de temperatura. Este proceso se conoce con el nombre de desnaturalización y sucede a valores de pH próximos a 13 o temperaturas alrededor de 100 ºC .
La temperatura a la que un 50% de la doble hélice se encuentra separada se conoce como temperatura de fusión (Tm); su valor difiere de unas muestras de DNA a otras y está en función del contenido en pares G-C. Esto es debido a que, al estar los pares G-C unidos por tres puentes de hidrógeno frente a los dos de los pares A-T, es necesaria una mayor cantidad de energía para desestabilizar una doble hélice rica en pares G-C. Así, la temperatura de fusión de una muestra de DNA es un indicador de su composición en bases nitrogenadas.
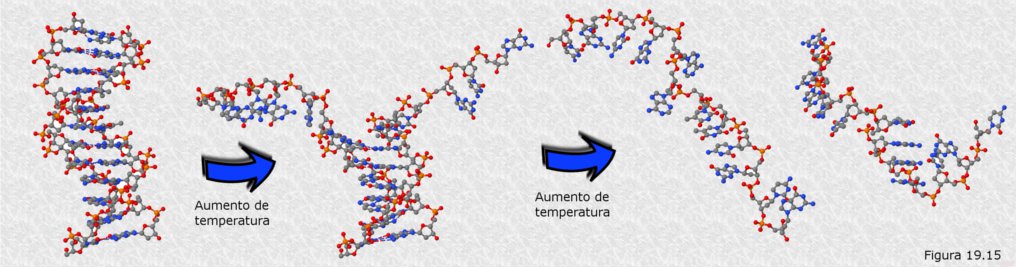
La desnaturalización del DNA (Figura 19.15) puede seguirse experimentalmente midiendo en un espectrofotómetro la absorción de luz ultravioleta por la disolución que lo contiene. La absorción de luz ultravioleta aumenta considerablemente con la desnaturalización debido a que los anillos aromáticos de las bases nitrogenadas absorben mucha más luz de esa longitud de onda cuando se encuentran desplegadas que cuando están apiladas en el interior de la doble hélice.
De manera análoga a lo que sucede con las proteínas, la desnaturalización del DNA es, en determinadas condiciones experimentales, reversible, siendo este proceso conocido como renaturalización. Si las cadenas polinucleotídicas que resultan de la desnaturalización se incuban a unos 65 ºC durante varias horas, se comprueba que las dobles hélices originales se reconstruyen espontáneamente. Paralelamente se produce el consiguiente descenso en la absorción de luz ultravioleta.
La renaturalización sirvió de base para el desarrollo de las llamadas técnicas de hibridación del DNA. Si se mezclan muestras de DNA desnaturalizado procedentes de especies diferentes y se incuba la mezcla en condiciones adecuadas para que se produzca la renaturalización, una fracción de las dobles hélices obtenidas serán híbridas, es decir, con cadenas polinucleotídicas de una y otra especie. El porcentaje de hibridación será mayor cuanto más parecidas sean las secuencias de nucleótidos de ambas especies, de manera que este porcentaje puede utilizarse como un indicador del parentesco evolutivo existente entre ellas. Antes de que estuviesen disponibles las actuales técnicas de secuenciación del DNA se utilizaron con profusión las técnicas de hibridación para el análisis de dicho parentesco.
3.- EL RNA: ESTRUCTURA Y TIPOS.
Como ya se ha comentado con anterioridad, el conocimiento de la química elemental de los ácidos nucleicos se demoró hasta la década de los años 20 del S XX. En la década siguiente fue reconocida por P. A. Levene la existencia de dos tipos diferentes de ácido nucleico (DNA y RNA) que diferían en algunos de sus componentes moleculares (el RNA incluía ribosa y uracilo en lugar respectivamente de la desoxirribosa y la timina del DNA). En esta misma época se realizaron estudios citológicos, usando colorantes y reactivos químicos específicos, para determinar la localización intracelular de uno y otro tipo de ácido nucleico. Se comprobó que en las células eucariotas la casi totalidad del DNA celular se encuentra en el interior del núcleo mientras que la mayor parte del RNA se encuentra en el citoplasma (aunque algunas zonas del núcleo, en particular el nucléolo, también son ricas en RNA). Por otra parte, del total de RNA citoplasmático una fracción muy importante se encontraba asociado a determinadas proteínas para formar unas partículas, visibles al microscopio electrónico, que fueron denominadas ribosomas. Experimentos realizados utilizando aminoácidos marcados radiactivamente pronto demostraron que los ribosomas eran el lugar de la célula donde se llevaba a cabo la síntesis de las proteínas, por lo que ya desde entonces se asoció al RNA con este proceso. Sin embargo, hubieron de pasar todavía algunos años hasta que se comprendió cual es el papel concreto que el RNA desempeña en el mismo.
3.1.- ESTRUCTURA Y FUNCIÓN DEL RNA.
La estructura tridimensional del RNA difiere claramente de la del DNA. En general las moléculas de RNA son monocatenarias (una sola cadena polinucleotídica), por lo que su composición en bases nitrogenadas no se ajusta a la regla de equivalencia de Chargaff. Sin embargo, existen moléculas de RNA que, aun siendo monocatenarias, presentan tramos con secuencias de bases complementarias los cuales adoptan estructuras en doble hélice, denominadas horquillas, de características análogas a las del DNA. En ocasiones, cuando las secuencias complementarias no son contiguas, se forman bucles de bases no emparejadas dentro de las horquillas. En las dobles hélices de RNA la adenina se empareja con el uracilo, que tiene estructura similar e idénticas posibilidades de formar puentes de hidrógeno que la timina, y la guanina con la citosina.
Hoy sabemos que la función primordial del RNA en las células consiste en servir de intermediario para transferir la información genética cifrada en el DNA a la estructura tridimensional de las proteínas en el proceso de expresión de la información genética, que analizaremos más adelante en este capítulo. De todos modos, en algunos virus el RNA constituye en sí mismo el material genético además de servir de intermediario en el proceso de síntesis de las proteínas virales. En algunos de estos virus la molécula de RNA que constituye el cromosoma viral es bicatenaria y presenta en toda su longitud estructura de doble hélice. También existen virus con cromosomas de RNA monocatenario.
3.2.- TIPOS DE RNA.
Existen varios tipos de RNA que difieren en el tamaño, estructura y función específica de sus moléculas. Todos ellos se sintetizan en el núcleo celular a partir de secuencias de DNA que sirven como molde y una vez sintetizados atraviesan los poros nucleares y se incorporan a sus diferentes destinos en el citoplasma.
a) RNA ribosómico (rRNA).- Constituye alrededor del 80% del RNA celular total. Sus moléculas son monocatenarias y de gran longitud (varios miles de nucleótidos). Algunas de ellas presentan horquillas y bucles con secuencias internas complementarias. Se encuentra en el citoplasma donde, en asociación con diferentes proteínas, forma parte estructural de los ribosomas.
b) RNA de transferencia (tRNA).- Sus moléculas son relativamente pequeñas (75 a 90 nucleótidos de longitud). Presentan una estructura característica con horquillas y bucles que les dan el aspecto de hojas de trébol cuando se representan sobre un plano: su estructura tridimensional presenta en realidad forma de L invertida. El tRNA presenta bases nitrogenadas diferentes a las características de los ácidos nucleicos en una proporción que puede alcanzar el 10% del total. Su función consiste en transportar de manera específica a los diferentes aminoácidos hasta los ribosomas para que allí sean ensamblados en las cadenas polipeptídicas en formación. Existen alrededor de 50 tipos de tRNAs que difieren en sus secuencias de nucleótidos y en algunos aspectos de su conformación tridimensional; sin embargo todos ellos comparten algunas características:
· En el extremo 5’ de la cadena polinucleotídica hay un triplete de bases nitrogenadas una de las cuales es siempre guanina.
· En el extremo 3’ la cadena polinucleotídica finaliza con la secuencia CCA y estas bases no están emparejadas. En este lugar es donde el tRNA se une a su aminoácido correspondiente.
· La molécula presenta tres brazos cada uno de los cuales consta de una horquilla con estructura en doble hélice y un bucle formado por bases sin emparejar (Figura 19.16). Se distinguen el brazo T (por donde la molécula se une al ribosoma), el brazo D (lugar que reconocen los enzimas específicos que unen los tRNA con sus aminoácidos correspondientes) y el brazo A (cuyo bucle presenta un triplete de bases, denominado anticodon, que es complementario de otro triplete, llamado codon, que se encuentra en el RNA mensajero, siendo esta complementariedad de gran importancia en el proceso de síntesis de proteínas).
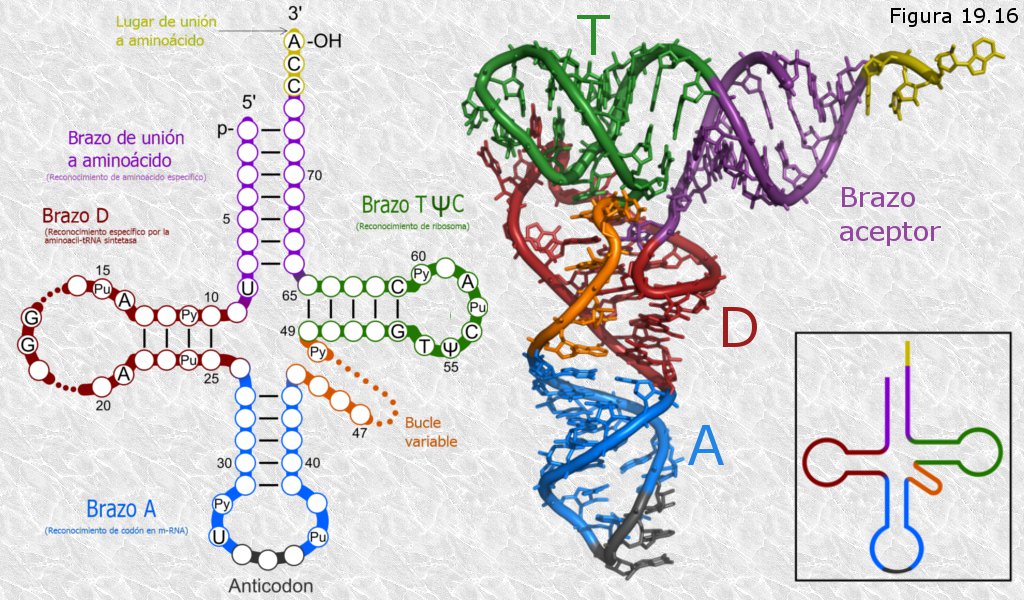
c) RNA mensajero (mRNA).- Sus moléculas están formadas en general por varios miles de nucleótidos y tienen una estructura lineal, aunque en ocasiones presentan horquillas y bucles. Su misión es trasladar la información genética almacenada en el DNA hasta los ribosomas para que allí se exprese en forma de proteínas. Los mRNA son en general moléculas de vida muy corta, ya que una vez cumplida su misión son degradados por unos enzimas llamados ribonucleasas.
d) RNA nucleolar (nRNA).- Se encuentra en el nucléolo, donde tiene lugar su síntesis. Una vez sintetizado se fragmenta por acción enzimática y da lugar a diferentes tipos de rRNA y tRNA.
4.- LA REPLICACIÓN DEL DNA.
Cada vez que una
célula se reproduce por división su material genético debe ser copiado
para que las dos células hijas dispongan una copia completa del mismo.
Uno de los requisitos que debería cumplir una eventual molécula de la
herencia, según apuntaba Schrödinger, es precisamente la capacidad para
crear copias fieles de sí misma de manera que la información pudiese ser
transmitida en las sucesivas generaciones celulares. Watson y Crick se
percataron, y así lo sugerían en su artículo de Nature, de que el
modelo de estructura en doble hélice que habían elaborado proporcionaba
las bases físico-químicas para un mecanismo de replicación del DNA. En
efecto, la estricta complementariedad de bases nitrogenadas a lo largo
de la doble hélice permite que cada una de las dos cadenas
polinucleotídicas que la forman pueda ser utilizada como molde
para sintetizar una nueva cadena complementaria. Así, desenrollando las
dos cadenas de una hélice original y utilizando cada una de ellas como
molde para sintetizar una nueva cadena, en la que los nucleótidos se
irán añadiendo conforme a las reglas que rigen el emparejamiento de las
bases, se obtendrán dos dobles hélices “hijas” idénticas, portadoras de
la misma información. El mecanismo que aquí se ha esbozado fue propuesto
por Watson y Crick en un artículo publicado en el número de Nature
de mayo de 1953, aclarando así lo que sugerían en su artículo del mes
anterior. Tal mecanismo fue conocido como replicación
semiconservativa para resaltar el hecho de que cada una de las
dobles hélices hijas conserva la mitad, es decir, una de las cadenas, de
la doble hélice original.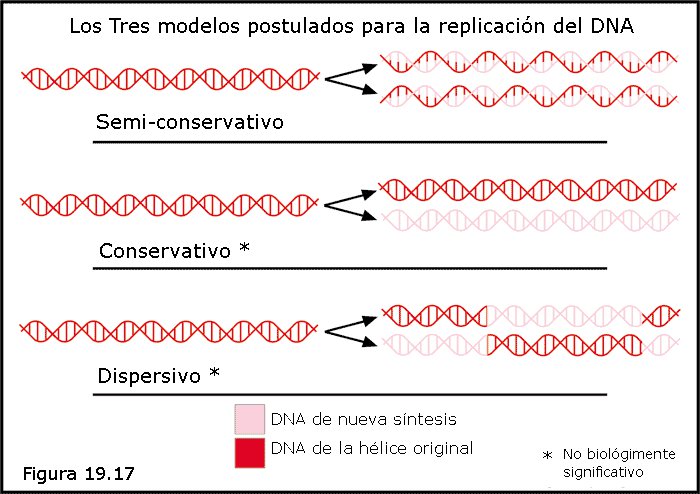
El modelo de replicación semiconservativa propuesta por Watson y Crick pareció acertado a muchos investigadores por su sencillez y elegancia. Sin embargo, se plantearon algunos modelos alternativos que no podían ser descartados a priori (Figura 19.17). Uno de ellos era el modelo de replicación conservativa, según el cual la doble hélice original, sin perder su integridad, sirve de patrón para sintetizar una doble hélice hija totalmente nueva, de manera que la doble hélice original se conserva intacta a lo largo de las sucesivas generaciones celulares. Otro era el modelo de replicación dispersiva, según el cual la doble hélice original se descompone en infinidad de pequeños fragmentos, cada uno de los cuales sirve de molde para sintetizar un fragmento complementario siguiendo las reglas de emparejamiento de bases; a continuación los fragmentos resultantes se empalman en el orden correcto para dar lugar a dos dobles hélices hijas cada una de las cuales contiene fragmentos de la original y fragmentos de nueva síntesis.
El modelo conservativo fue propuesto por algunos investigadores a la vista de algunas dificultades técnicas que parecía presentar el modelo semiconservativo. Estos investigadores eran de la opinión de que la doble hélice del DNA se replicaba de una manera indirecta, transfiriendo primero su información a un intermediario de otra naturaleza (probablemente una proteína), que serviría después de patrón para sintetizar una doble hélice totalmente nueva. Por otra parte, el modelo dispersivo gozaba de muy pocos partidarios, ya que la correcta ordenación de los fragmentos de DNA requeriría la presencia de una molécula distinta del propio DNA que poseyese la información necesaria para ordenarlos; ello sería equivalente a afirmar que esta molécula, y no el DNA, era en realidad el material genético, razón por la cual este modelo sólo tenía un cierto apoyo entre los pocos investigadores que seguían atribuyendo este papel a las proteínas.
4.1.- EL EXPERIMENTO DE MESELSON Y STAHL.
 Un brillante
experimento realizado por M. S. Meselson y F. W. Stahl
(Figura 19.18) en 1957 demostró
que el modelo semiconservativo propuesto por Watson Y Crick era el
correcto. El diseño experimental se basó en el uso de dos isótopos
estables del nitrógeno: el isótopo más abundante en la naturaleza (14N)
y un isótopo pesado (15N), más escaso. En primer lugar
Meselson y Stahl dispusieron dos cultivos de la bacteria E. coli
creciendo sobre un medio en el que la única fuente de nitrógeno era NH4Cl.
En uno de los cultivos el NH4Cl contenía el isótopo normal
del nitrógeno (14N)
y en el otro el isótopo pesado (15N). De este modo, el
isótopo correspondiente a cada medio de cultivo se incorporaba a todas
las biomoléculas nitrogenadas de las bacterias que crecían en él,
incluyendo los ácidos nucleicos. A continuación extrajeron el DNA de las
células de uno y otro cultivo, lo mezclaron y lo sometieron a
centrifugación a alta velocidad en un gradiente de densidad de CsCl. Así
comprobaron que el DNA “ligero” y el DNA “pesado” que habían incorporado
uno u otro isótopo se situaban en dos bandas perfectamente distinguibles
en el tubo de la centrífuga (Figura
19.20).
Un brillante
experimento realizado por M. S. Meselson y F. W. Stahl
(Figura 19.18) en 1957 demostró
que el modelo semiconservativo propuesto por Watson Y Crick era el
correcto. El diseño experimental se basó en el uso de dos isótopos
estables del nitrógeno: el isótopo más abundante en la naturaleza (14N)
y un isótopo pesado (15N), más escaso. En primer lugar
Meselson y Stahl dispusieron dos cultivos de la bacteria E. coli
creciendo sobre un medio en el que la única fuente de nitrógeno era NH4Cl.
En uno de los cultivos el NH4Cl contenía el isótopo normal
del nitrógeno (14N)
y en el otro el isótopo pesado (15N). De este modo, el
isótopo correspondiente a cada medio de cultivo se incorporaba a todas
las biomoléculas nitrogenadas de las bacterias que crecían en él,
incluyendo los ácidos nucleicos. A continuación extrajeron el DNA de las
células de uno y otro cultivo, lo mezclaron y lo sometieron a
centrifugación a alta velocidad en un gradiente de densidad de CsCl. Así
comprobaron que el DNA “ligero” y el DNA “pesado” que habían incorporado
uno u otro isótopo se situaban en dos bandas perfectamente distinguibles
en el tubo de la centrífuga (Figura
19.20).
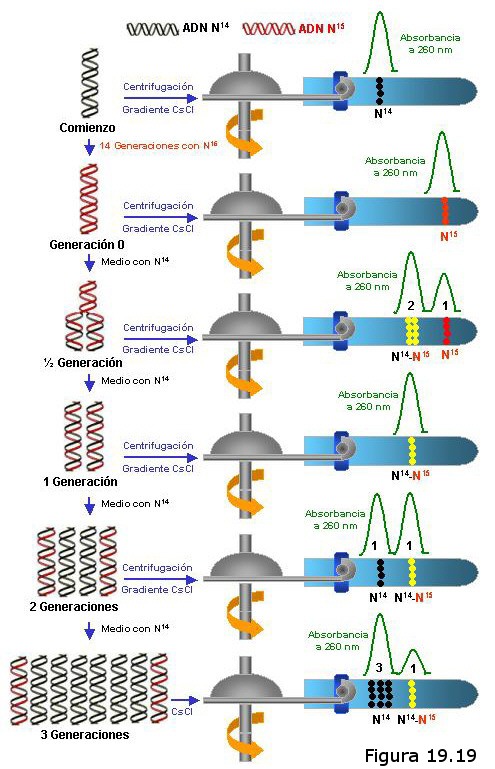
En una segunda fase de su experimento Meselson y Stahl transfirieron bacterias que habían estado reproduciéndose durante varias generaciones en un medio de cultivo con nitrógeno pesado a un medio de cultivo con nitrógeno ligero, de manera que, a partir del instante de la transferencia, todo el DNA que se sintetizase en las células transferidas incorporaría exclusivamente nitrógeno ligero. Seguidamente, a intervalos regulares de 20 minutos (equivalentes al período de generación de las bacterias) extrajeron el DNA de sucesivas muestras del cultivo y lo sometieron a centrifugación en gradiente de densidad de CsCl.
 El éxito del experimento de Meselson y
Stahl radicó en que su diseño permitía formular predicciones sobre los
resultados en función de cada uno de los tres modelos de replicación
propuestos, siendo los resultados predichos perfectamente distinguibles
y excluyentes entre sí.
El éxito del experimento de Meselson y
Stahl radicó en que su diseño permitía formular predicciones sobre los
resultados en función de cada uno de los tres modelos de replicación
propuestos, siendo los resultados predichos perfectamente distinguibles
y excluyentes entre sí.
En efecto, si el modelo de replicación fuese semiconservativo como habían predicho Watson y Crick, todo el DNA extraído tras los 20 primeros minutos consistiría en dobles hélices formadas por una cadena ligera (recién sintetizada) y otra cadena pesada (la original), lo que se traduciría en un DNA de densidad “híbrida” que en el tubo de la centrífuga ocuparía una posición intermedia entre las correspondientes al DNA “pesado” y el “ligero”. Transcurridos otros 20 minutos, es decir, en la segunda generación de bacterias tras la transferencia, la mitad de las dobles hélices serían de densidad híbrida mientras que la otra mitad serían totalmente ligeras, dando lugar en el tubo de la centrífuga a dos bandas en las posiciones correspondientes.
Si por el contrario la replicación respondiese a un modelo conservativo nunca aparecerían bandas híbridas, pues el DNA de las bacterias de la primera generación estaría compuesto por dobles hélices la mitad de las cuales serían totalmente ligeras (las recién sintetizadas) y la otra mitad totalmente pesadas (las originales), de manera que en el tubo de la centrífuga aparecería una banda ligera y otra pesada. Lo mismo ocurriría en la segunda generación de bacterias, con la única diferencia de que la proporción de hélices ligeras respecto a hélices pesadas sería de 3:1 en lugar de 1:1, lo que se traduciría en una mayor densidad de moléculas de DNA ligero en la banda correspondiente.
En el caso de que la replicación siguiese un modelo dispersivo los resultados obtenidos en la primera generación serían iguales a los que predice el modelo semiconservativo: el DNA extraído de las bacterias de la primera generación estaría formado por fragmentos ligeros y fragmentos pesados que se repartirían en una y otra cadena polinucleotídica de la doble hélice, y ello daría lugar a la aparición de una única banda en el tubo de la centrífuga que ocuparía la posición correspondiente al DNA de densidad híbrida. Esta banda híbrida persistiría en la segunda y sucesivas generaciones, aunque cabría esperar que tras varios ciclos de replicación, al ir aumentando la proporción de fragmentos ligeros con respecto a los pesados, empezasen a aparecer dobles hélices totalmente ligeras que irían formando una banda en su posición correspondiente.
Los resultados obtenidos por Meselson y Stahl se ajustaban con precisión a los predichos por el modelo de replicación semiconservativa. No obstante, dado que también eran parcialmente compatibles con el modelo dispersivo (al menos en lo que se refiere a las bandas obtenidas con el DNA de la primera generación), realizaron una posterior comprobación con el objeto de descartar definitivamente este modelo. Tomaron una nueva muestra de DNA de las bacterias de la primera generación y, antes de centrifugarlo, procedieron a desnaturalizarlo. Así, al separar físicamente las dos cadenas polinucleotídicas que integran la doble hélice deberían obtenerse dos bandas (una ligera y otra pesada) en el tubo de la centrífuga en el caso de que la replicación fuese semiconservativa, o bien una sola banda híbrida en el caso de que fuese dispersiva. Los resultados de este último experimento no dejaban lugar a dudas: Watson y Crick habían acertado también con el mecanismo que rige la replicación del DNA.
4.2.- OTRAS CARACTERÍSTICAS DE LA REPLICACION.
Una vez demostrado el carácter semiconservativo de la replicación del DNA se plantearon nuevos interrogantes acerca de algunos aspectos del proceso. En primer lugar cabe preguntarse si en la replicación de una gran molécula de DNA ¿se desenrollan y separan primero las dos cadenas polinucleotídicas en toda su longitud para luego servir cada una de ellas como molde para la síntesis de su complementaria?, o bien ¿los procesos de separación de las cadenas y la síntesis de sus complementarias están de alguna manera acopladas en el espacio y en el tiempo?
Los estudios realizados por John Cairns y otros investigadores en la década de los años 60 demostraron que la replicación del DNA es ordenada y secuencial, es decir, se inicia en unos puntos fijos del cromosoma, y el crecimiento de las nuevas cadenas de DNA se produce de manera simultánea al desenrollamiento de la doble hélice original.
Para elucidar este y otros aspectos de la replicación del DNA, Cairns utilizó una técnica basada en isótopos radiactivos denominada autorradiografía. En un cultivo de E. coli introdujo en el medio de cultivo timina tritiada (timina en la que los átomos de hidrógeno habían sido sustituidos por su isótopo radiactivo 3H), de manera que esta base nitrogenada se incorporaba a las cadenas de DNA en crecimiento. Transcurridas dos generaciones procedió a la extracción del DNA bacteriano mediante una técnica que respetaba la integridad de las moléculas extraídas (lisis de la pared celular con enzimas específicos) y permitió que estas moléculas sedimentasen lentamente sobre un papel de filtro que después fue cuidadosamente montado sobre una placa con emulsión fotosensible. La placa se guardó en la oscuridad durante dos meses y a continuación se procedió a su revelado. Las emisiones β de los átomos de tritio incorporados al DNA en los restos de timina habían impresionado la placa fotográfica creando unas figuras que se correspondían con cromosomas bacterianos en distintos estadios de replicación (Figura 19.20). El análisis de estas imágenes, además de proporcionar la primera prueba directa del carácter circular del cromosoma bacteriano, condujo a la conclusión de que la replicación comienza en un único origen, en el que se forma una única “burbuja de replicación” que va creciendo a medida que el proceso avanza.
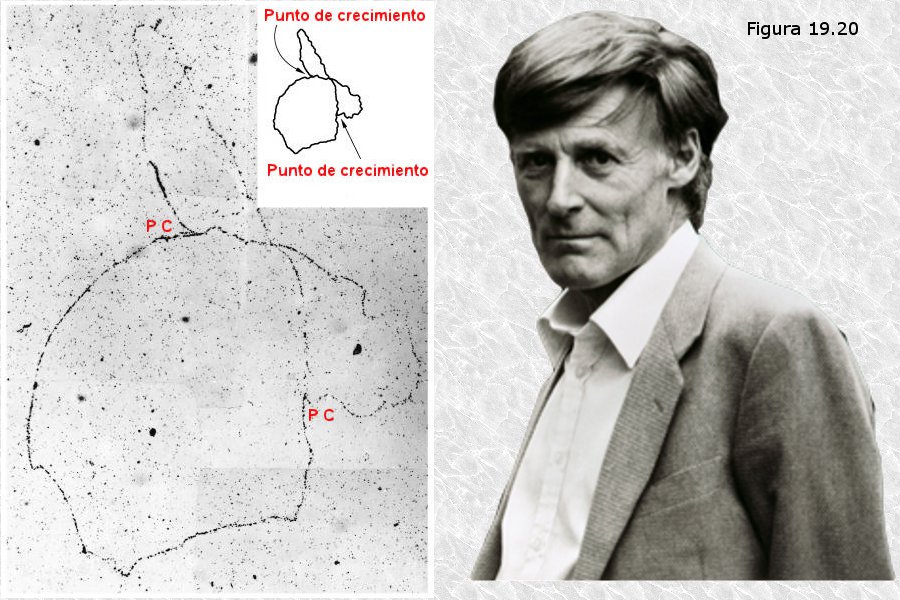
Posteriores refinamientos de la técnica de autorradiografía, consistentes en el uso de “pulsos” de timina tritiada de pocos segundos de duración, permitieron obtener imágenes de las zonas del cromosoma en las que está teniendo lugar la replicación del DNA en un instante dado. El análisis de estas imágenes reveló que el proceso de replicación es bidireccional: dos horquillas de replicación avanzan simultáneamente en direcciones opuestas partiendo de un único origen.
Años más tarde, usando las mismas técnicas, se pudo comprobar que en los cromosomas eucariotas la replicación se atiene a los mismos principios que en los procariotas, con la excepción de que en aquéllos existen varios orígenes de replicación que dan lugar a varios ojos de replicación por cromosoma. Estos ojos de replicación crecen de manera bidireccional hasta que se funden con sus vecinos completando el proceso.
4.3.- SÍNTESIS ENZIMÁTICA DEL DNA.
El proceso de replicación del DNA implica la adición de nucleótidos a una cadena polinucleotídica en crecimiento complementaria de la que le sirve como molde. Los sucesivos nucleótidos deben establecer enlaces fosfodiéster con los restos precedentes y la formación de estos enlaces requiere energía. Debido a ello, los nuevos nucleótidos que se incorporan al proceso no pueden ser nucleótidos monofosfato sino que las células deben recurrir a nucleótidos trifosfato, mucho más energéticos, cuya escisión pirofosforolítica proporciona la energía necesaria para la formación de dichos enlaces. La incorporación de un resto nucleotídico a la cadena en crecimiento supone pues la siguiente reacción química:
DNA (n) + dNTP à DNA (n+1) + PPi
 Es conveniente resaltar que esta reacción química, como la
mayoría de las que ocurren en las células vivas, es reversible, por lo
que altas concentraciones de pirofosfato inorgánico podrían hacer
revertir esta reacción hacia la degradación (pirofosforólisis) del DNA
celular, con la consiguiente muerte de la célula. Por ello, las células
vivas disponen de un enzima de gran eficacia y ubicuidad, llamado
pirofosfatasa inorgánica, que degrada rápidamente el pirofosfato (PPi)
a ortofosfato (2 Pi) con el objeto de preservar la integridad
de los ácidos nucleicos celulares a pesar del considerable gasto
energético que esta degradación supone.
Es conveniente resaltar que esta reacción química, como la
mayoría de las que ocurren en las células vivas, es reversible, por lo
que altas concentraciones de pirofosfato inorgánico podrían hacer
revertir esta reacción hacia la degradación (pirofosforólisis) del DNA
celular, con la consiguiente muerte de la célula. Por ello, las células
vivas disponen de un enzima de gran eficacia y ubicuidad, llamado
pirofosfatasa inorgánica, que degrada rápidamente el pirofosfato (PPi)
a ortofosfato (2 Pi) con el objeto de preservar la integridad
de los ácidos nucleicos celulares a pesar del considerable gasto
energético que esta degradación supone.
La reacción de polimerización del DNA, como todas las reacciones químicas celulares requiere de la actuación de enzimas específicos que catalicen y controlen este proceso. A mediados de los años 50 del S.XX, en paralelo con los estudios realizados acerca del mecanismo de la replicación, el equipo Arthur Kornberg (Figura 19.21) inició sus investigaciones acerca de la síntesis enzimática del DNA en la bacteria E. coli. Pronto consiguió identificar y aislar un enzima capaz de catalizar la reacción de polimerización que fue denominado DNA polimerasa I. Estudios posteriores pusieron de manifiesto que el proceso de replicación presentaba una gran complejidad y que era necesaria la concurrencia de toda una batería de enzimas y otras proteínas no enzimátitcas para que culminase con éxito. Hoy, tras 50 años de estudios, se tiene un amplio conocimiento acerca de todo este complejo proceso y de los enzimas implicados, tanto en lo tocante a las células procariotas como, aunque en menor medida, a las eucariotas.
A continuación se describe el equipo de enzimas y proteínas no enzimáticas implicados en la síntesis del DNA con sus características más relevantes:
-
Proteínas DNA A (producto del gen del mismo nombre).- Relajan la doble hélice en la zona donde ha de iniciarse la replicación preparándola para la acción de las siguientes proteínas.
-
Helicasas.- Son de varios tipos (proteínas B, C, J, K). Su actividad consiste en desenrollar las dos cadenas de la doble hélice haciéndolas girar alrededor del eje central. En el inicio del proceso dan lugar a la llamada “burbuja” u “ojal” de replicación. A partir del origen dos equipos de helicasas se desplazan en sentidos opuestos haciendo avanzar las dos horquillas de replicación.
-
Proteínas SSB (single strand binding).- Moléculas que se unen en gran número a las cadenas sencillas de DNA y las estabilizan con el objeto de evitar que vuelvan a asociarse y que se cierre así la burbuja de replicación.
-
Girasas y topoisomerasas.- El desenrollamiento de la doble hélice por acción de las helicasas genera tensiones en la molécula de DNA que tiende a formar “superenrollamientos” del tipo de los que aparecen en el cable de un auricular telefónico cuando inadvertidamente lo hacemos girar sobre sí mismo. Para relajar estas tensiones unos enzimas llamados topoisomerasas cortan de trecho en trecho algún enlace fosfodiéster de la cadena molde y lo vuelven a soldar después de que otras proteínas, las girasas, liberan la tensión mediante el giro del extremo que queda temporalmente libre.
-
Exonucleasas.- Enzimas que escinden nucleótidos a partir del extremo de una cadena polinucleotídica. Unas actúan en dirección 5’à3’ y otras lo hacen en dirección 3’à5’. Su función es eliminar fragmentos de cadenas polinucleotídicas en algunas fases del proceso de replicación.
-
DNA Polimerasas.- Enzimas que añaden nucleótidos a una cadena de DNA en crecimiento que es complementaria de otra que utilizan como molde. Existen varios tipos de DNA polimerasas que difieren en sus características y actividades. En células procariotas se han identificado 3 DNA polimerasas (I, II y III) mientras que en células eucariotas se han identificado varias (α, β, γ, δ, ε). Todas las DNA polimerasas conocidas añaden nucleótidos a una cadena que crece en dirección 5’à3 recorriendo el molde en dirección 3’à5’, es decir, catalizan la condensación del grupo 3’ OH libre del último nucleótido de la cadena con el grupo α-5’fosfato del nucleótido que se añade. Por otra parte, todas necesitan de una cadena que actúe como molde, es decir, no pueden añadir nucleótidos sin más a una cadena polinucleotídica simple. Otro rasgo importante de todas las DNA polimerasas es que todas ellas pueden prolongar una cadena polinucleotídica pero ninguna de ellas puede iniciarlas, es decir, no pueden colocar el primer nucleótido de la cadena sino que precisan de un tramo preexistente, que en lo sucesivo llamaremos primer o cebador, al que añadir nuevos nucleótidos.
-
Primasas.- Enzimas capaces de sintetizar una cadena corta de RNA utilizando un molde de DNA (en realidad son RNA polimerasas-DNA dirigidas). A diferencia de las DNA polimerasas, las primasas sí pueden iniciar cadenas. Son las encargadas de crear los cebadores de RNA sobre los que después han de actuar las DNA polimerasas.
-
DNA Ligasas.- Enzimas que establecen puentes fosfodiéster entre nucleótidos adyacentes que se encuentran en fragmentos diferentes de cadena polinucleotídica. Son necesarias en distintos momentos del proceso replicativo para “soldar” los fragmentos sueltos.
Además de los descritos existen algunos enzimas mixtos, que en una sola molécula presentan más de una actividad. Por ejemplo, las DNA polimerasas presentan en general actividad de exonucleasa en una o en ambas direcciones de la cadena.
A continuación analizaremos el proceso de replicación del DNA deteniéndonos en el papel que desempeña cada uno de los distintos tipos de enzimas y proteínas no enzimáticas implicadas y en la secuencia temporal de su actuación. Por ser el más estudiado, adoptaremos como modelo el proceso de replicación en la bacteria E. coli, que con muy pequeñas modificaciones es extensible a todas las células procariotas. Seguidamente se analizarán algunas particularidades que presenta el proceso replicativo en las células eucariotas. Distinguiremos en el proceso tres fases: iniciación, elongación y terminación.
FASE DE INICIACIÓN.
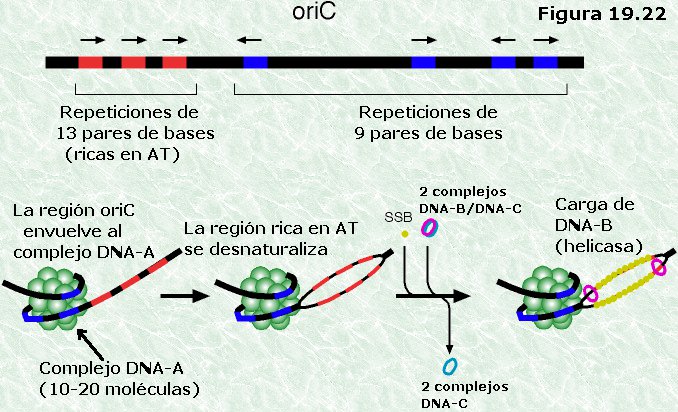 La replicación del DNA se inicia en un lugar concreto del
cromosoma denominado origen de replicación. Existe un único
origen por cromosoma en todas las especies bacterianas conocidas. El
origen de replicación consiste en un tramo de 245 pares de base de
longitud, conocido como oriC, que contiene varias repeticiones en
serie de secuencias específicas de nucleótidos que han podido ser
identificadas. Estas secuencias son ricas en pares A-T, lo que facilita
la apertura de la doble hélice, y son reconocibles por proteínas
específicas que se fijan a ellas.
La replicación del DNA se inicia en un lugar concreto del
cromosoma denominado origen de replicación. Existe un único
origen por cromosoma en todas las especies bacterianas conocidas. El
origen de replicación consiste en un tramo de 245 pares de base de
longitud, conocido como oriC, que contiene varias repeticiones en
serie de secuencias específicas de nucleótidos que han podido ser
identificadas. Estas secuencias son ricas en pares A-T, lo que facilita
la apertura de la doble hélice, y son reconocibles por proteínas
específicas que se fijan a ellas.
Un ciclo de replicación comienza cuando varias unidades monoméricas de la proteína específica DNA A se fijan sobre las correspondientes secuencias específicas repetidas de 9 pares de bases de longitud dentro del tramo oriC (Figura 19.22). A continuación se van añadiendo nuevas unidades monoméricas de DNA A que forman un complejo nucleoproteico con unas veinte de estas unidades y el tramo oriC de la doble hélice de DNA que se dispone formando un bucle alrededor de ellas. Algunos de los monómeros de DNA A quedan así situados sobre las secuencias específicas repetitivas de 13 pares de bases de longitud que se encuentran en el otro extremo de oriC, provocando en ellas una desestabilización de la doble hélice y la consiguiente apertura de la burbuja de replicación. La proteína DNA A tiene la función de reconocer el origen de replicación, provocar la apertura de la burbuja y facilitar la entrada de las demás proteínas que forman parte del complejo de iniciación.
Una vez abierta la burbuja de replicación se introducen en ella las proteínas denominadas DNA B y DNA C. La primera de ellas tiene actividad helicasa y actúa agrandando la burbuja de replicación (dos moléculas de DNA B actúan en direcciones opuestas haciendo avanzar las recién creadas horquillas). La presencia de DNA C se requiere únicamente para que DNA B inicie su actuación, abandonando el complejo inmediatamente después. Por el contrario, la helicasa DNA B, a la que posteriormente se unen las helicasas DNA J y DNA K, permanece en su lugar y sigue actuando también durante la fase de elongación.
A medida que la burbuja de replicación se va haciendo mayor van quedando expuestos tramos cada vez mayores de DNA de cadena sencilla. A estos tramos se unen las proteínas SSB con el objeto de protegerlos del ataque de nucleasas que podrían degradarlos y de evitar que se reconstruya la doble hélice cerrándose con ello la propia burbuja de replicación.
Dado que no existe ninguna DNA polimerasa capaz de iniciar cadenas polinucleotídicas se hace necesaria ahora la concurrencia de una primasa (proteína DNA G) que inicia una cadena corta de RNA (unos 12 nucleótidos de longitud) que actuará como cebador al que la DNA polimerasa podrá añadir nucleótidos en la fase de elongación (Figura 19.25). La primasa DNA G junto con la helicasa DNA B forman una unidad funcional denominada primosoma cuya integridad es necesaria para que la iniciación tenga lugar.
FASE DE ELONGACIÓN.

La fase de elongación comienza con la progresión en ambas direcciones de las horquillas de replicación mediante la acción de las helicasas, que van desenrollando la doble hélice. La acción combinada de girasa y topoisomerasa en ambas horquillas va relajando las tensiones asociadas al superenrollamiento. Mientras tanto, las proteínas SSB van estabilizando los tramos de cadena sencilla que quedan temporalmente expuestos. Todas estas actuaciones se prolongan durante toda la fase de elongación.
Es ahora cuando se pone de manifiesto un problema técnico
que afecta a todo el proceso de replicación y que las células vivas, en
el transcurso de su evolución, se han visto obligadas a solucionar. Dado
que las dos cadenas polinucleotídicas de una doble hélice son
antiparalelas, cuando una horquilla de replicación avanza, lo hace en la
dirección 5’à3’ para una de
las cadenas y en dirección 3’à5’
para la otra. Por otra parte, se ha comentado ya que todas las DNA
polimerasas conocidas añaden nucleótidos a una cadena que crece en
dirección 5’à3’ mientras que
recorren el molde en la dirección opuesta. Por todo ello, la síntesis de
DNA sólo puede acompañar al avance de una horquilla de replicación en
una de las cadenas (la que la DNA polimerasa recorre como molde
en dirección 3’à5’). ¿Qué
ocurre entonces con la otra cadena? El investigador japonés
R. Okazaki (Figura 19.23) demostró en 1968, utilizando el marcaje radiactivo con
timina tritiada, que la síntesis en esta otra cadena es
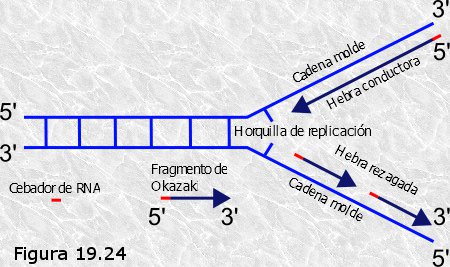 discontinua: las
DNA polimerasas incorporan nuevos nucleótidos en dirección
contraria a la de avance de la horquilla generando unos fragmentos de
DNA de 1.000 a 2.000 nucleótidos de longitud, denominados fragmentos
de Okazaki (Figura
19.24). La cadena en la que la síntesis es continua se denomina
hebra conductora (leader strand) y aquella en la que la
síntesis es discontinua se denomina hebra rezagada (lagging strand).
Como resulta evidente, el inicio de cada fragmento debe aguardar a que
la horquilla avance la longitud correspondiente para luego ir creciendo
en dirección contraria a dicho avance. Okazaki y sus colaboradores
demostraron en 1972 que cada fragmento recién formado lleva unido su
correspondiente cebador que deberá ser después eliminado.
discontinua: las
DNA polimerasas incorporan nuevos nucleótidos en dirección
contraria a la de avance de la horquilla generando unos fragmentos de
DNA de 1.000 a 2.000 nucleótidos de longitud, denominados fragmentos
de Okazaki (Figura
19.24). La cadena en la que la síntesis es continua se denomina
hebra conductora (leader strand) y aquella en la que la
síntesis es discontinua se denomina hebra rezagada (lagging strand).
Como resulta evidente, el inicio de cada fragmento debe aguardar a que
la horquilla avance la longitud correspondiente para luego ir creciendo
en dirección contraria a dicho avance. Okazaki y sus colaboradores
demostraron en 1972 que cada fragmento recién formado lleva unido su
correspondiente cebador que deberá ser después eliminado.
|
|
DNA pol I |
DNA pol II |
DNA pol III |
|
Actividad polimerasa
|
SI |
SI |
SI |
|
Actividad Exonucleasa 3’→5’
|
SI |
SI |
NO |
|
Actividad Exonucleasa 5’→3’
|
SI |
NO |
NO |
|
Función biológica |
Replicación DNA Eliminación cebadores |
Reparación DNA |
Replicación DNA |
| Tabla 19.1 | |||
En E. coli se han descubierto tres DNA polimerasas diferentes que poseen distintas combinaciones de actividad polimerasa y exonucleasa (Tabla 19.1). La actividad exonucleasa 3’à5’ tiene una función correctora: cuando se introduce un nucleótido incorrecto esta actividad lo elimina y el enzima se desplaza un lugar hacia atrás para introducir el correcto. Se ha podido comprobar que entre estos enzimas es la DNA polimerasa III la responsable principal del proceso replicativo, aunque la DNA polimerasa I también desempeña una importante función en el mismo. La DNA polimerasa II tiene una actividad relacionada con la reparación del DNA más que con la replicación.
Una vez colocados los dos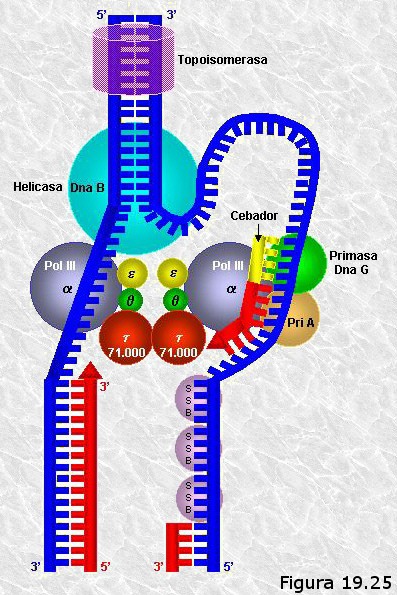 primeros cebadores por la
primasa, la DNA polimerasa III comienza a añadirles
nucleótidos en dirección 5’à3’
iniciando así las hebras conductoras de ambas horquillas de replicación.
Nos referiremos en lo sucesivo a lo que ocurre en una de estas
horquillas, considerando que los procesos que ocurren en la otra son
análogos. Cuando la hebra conductora ha avanzado entre 1.000 y 2.000
nucleótidos la primasa coloca el cebador del primer fragmento de
Okazaki, que a continuación es prolongado por la DNA polimerasa III
hasta que se encuentra con el cebador de la hebra conductora de la otra
horquilla de replicación y ahí se desprende. Este proceso se repite
cuantas veces sea necesario, a medida que el equipo de helicasas,
girasa y topoisomerasa va abriendo la doble hélice, hasta
completar la replicación del cromosoma. Un fragmento de Okazaki es
colocado en la hebra rezagada en cada ciclo mientras que la hebra
conductora se sintetiza sin interrupción; cada fragmento finaliza cuando
la DNA polimerasa III se encuentra con el cebador del fragmento
precedente, momento en que se disocia del DNA. En realidad la DNA
polimerasa III es una proteína oligomérica formada por múltiples
subunidades que se coloca en la horquilla de replicación de manera que
se encarga simultáneamente de la síntesis en las dos hebras merced a un
complejo mecanismo que implica la formación temporal de un lazo en la
hebra rezagada a medida que se va formando cada fragmento de Okazaki (Figura
19.25); este lazo desaparece cuando las subunidades del enzima
responsables de la síntesis del fragmento se disocian del DNA y se
vuelve a formar cuando dichas subunidades se vuelven a incorporar para
sintetizar el fragmento siguiente.
primeros cebadores por la
primasa, la DNA polimerasa III comienza a añadirles
nucleótidos en dirección 5’à3’
iniciando así las hebras conductoras de ambas horquillas de replicación.
Nos referiremos en lo sucesivo a lo que ocurre en una de estas
horquillas, considerando que los procesos que ocurren en la otra son
análogos. Cuando la hebra conductora ha avanzado entre 1.000 y 2.000
nucleótidos la primasa coloca el cebador del primer fragmento de
Okazaki, que a continuación es prolongado por la DNA polimerasa III
hasta que se encuentra con el cebador de la hebra conductora de la otra
horquilla de replicación y ahí se desprende. Este proceso se repite
cuantas veces sea necesario, a medida que el equipo de helicasas,
girasa y topoisomerasa va abriendo la doble hélice, hasta
completar la replicación del cromosoma. Un fragmento de Okazaki es
colocado en la hebra rezagada en cada ciclo mientras que la hebra
conductora se sintetiza sin interrupción; cada fragmento finaliza cuando
la DNA polimerasa III se encuentra con el cebador del fragmento
precedente, momento en que se disocia del DNA. En realidad la DNA
polimerasa III es una proteína oligomérica formada por múltiples
subunidades que se coloca en la horquilla de replicación de manera que
se encarga simultáneamente de la síntesis en las dos hebras merced a un
complejo mecanismo que implica la formación temporal de un lazo en la
hebra rezagada a medida que se va formando cada fragmento de Okazaki (Figura
19.25); este lazo desaparece cuando las subunidades del enzima
responsables de la síntesis del fragmento se disocian del DNA y se
vuelve a formar cuando dichas subunidades se vuelven a incorporar para
sintetizar el fragmento siguiente.
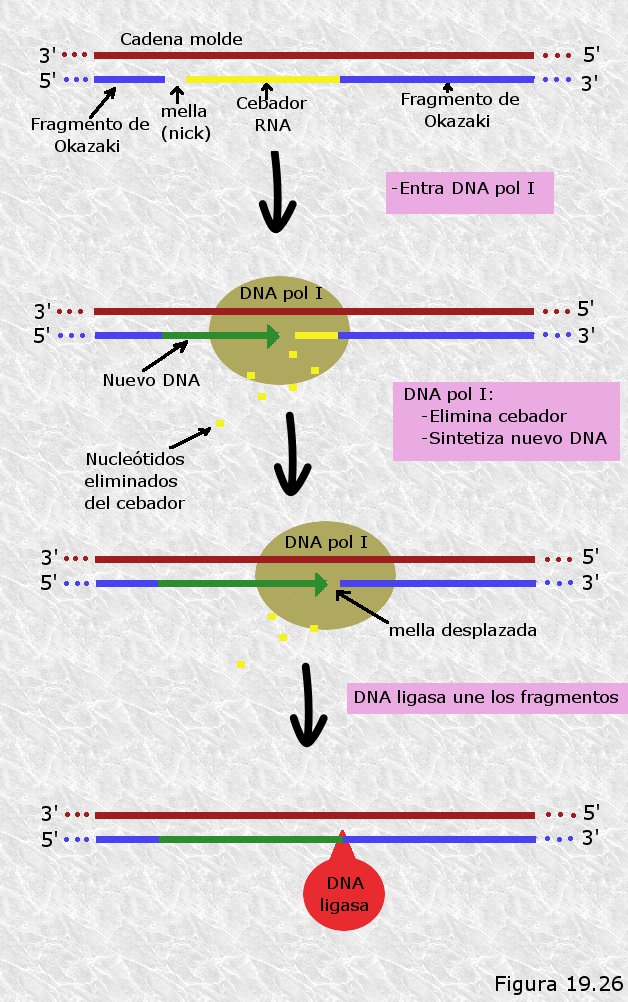
Del modo que se ha descrito hasta ahora, cuando las horquillas de replicación en su avance recorran todo el cromosoma bacteriano se habrá completado la replicación en las hebras conductoras mientras que las hebras rezagadas habrán quedado sembradas de fragmentos de Okazaki cada uno de ellos con su correspondiente cebador de RNA. Las tareas pendientes del equipo enzimático de la replicación consistirán ahora en eliminar los cebadores y sellar la unión de los sucesivos fragmentos de Okazaki. La eliminación de los cebadores es llevada a cabo por la DNA polimerasa I, enzima que, merced a la actividad exonucleasa 5’à3’ que lleva incorporada, puede alargar un fragmento dado y al tiempo ir degradando el cebador del fragmento precedente escindiendo uno a uno los ribonucleótidos que lo forman. Este proceso, conocido como corrimiento de mella (nick translation), sustituye el cebador por un tramo de DNA recién sintetizado de manera que la hebra rezagada consiste ahora en una sucesión de fragmentos de Okazaki formados exclusivamente por desoxirribonucleótidos (Figura 19.26). Así se eliminan todos los cebadores incluidos los de ambas hebras conductoras, que se ven afectados por la prolongación del primer fragmento de Okazaki sintetizado en las respectivas hebras rezagadas. Para completar la replicación sólo falta unir estos fragmentos y de ello se encarga un enzima específico, la DNA ligasa, que sella las uniones pendientes entre el último nucleótido de cada fragmento y el primero del siguiente, consumiendo para ello energía de la hidrólisis del ATP. Es conveniente resaltar que, si bien se han descrito como fenómenos separados en el tiempo para facilitar su comprensión, la eliminación de los cebadores y el sellado de los fragmentos de Okazaki discurren en realidad en paralelo con el avance de las horquillas de replicación.
FASE DE TERMINACIÓN.
Puesto que la replicación del cromosoma bacteriano comienza en un único origen y es bidireccional, las dos horquillas de replicación avanzando a velocidad constante se encontrarán en el lugar del cromosoma diametralmente opuesto al origen (oriC). Allí, las hebras conductoras de una y otra horquilla de replicación se detendrán al encontrarse con el cebador del último fragmento de Okazaki sintetizado en la hebra rezagada respectiva, que seguidamente será eliminado por la DNA polimerasa I y la mella resultante sellada por la DNA ligasa (Figura 19.27). No obstante lo dicho, se ha podido comprobar que el lugar de terminación de la replicación está señalizado por una secuencia específica de nucleótidos (el lugar Ter) a la que se fija una proteína específica que bloquea el avance de las helicasas cuando llegan a ella. Cuando se bloquea el avance de una de las horquillas en un lugar alejado del de terminación la otra horquilla se detiene al llegar a éste de todas maneras.
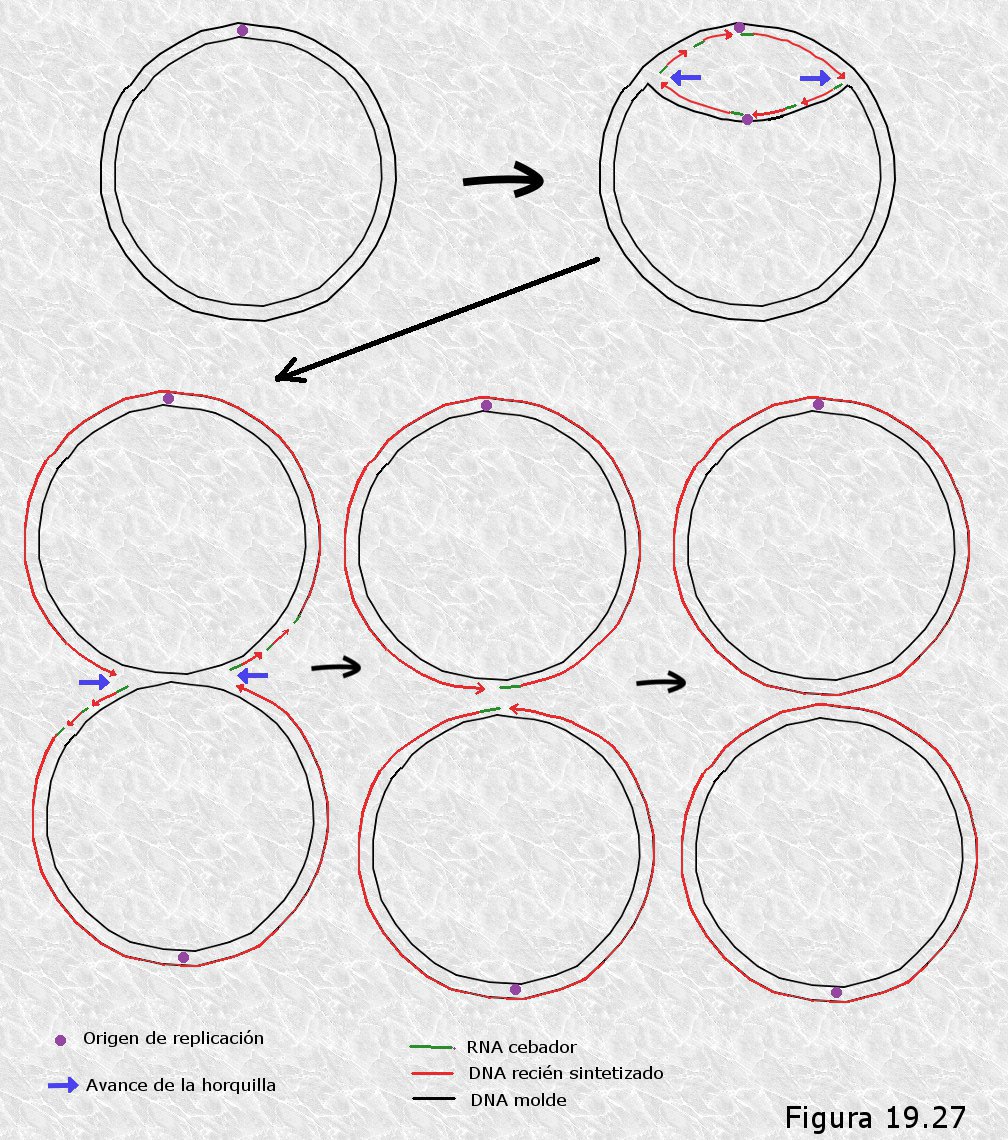
4.4.- REPLICACIÓN DEL DNA EN EUCARIOTAS.
La descripción hasta aquí realizada del proceso replicativo
en la bacteria E. coli es aplicable en líneas generales a todas
las células procariotas y en muchos aspectos también a las células
eucariotas. En ellas la replicación también es semiconservativa,
secuencial, bidireccional, continua en la hebra conductora y discontinua
en la rezagada, y es llevada a cabo por un equipo de enzimas y proteínas
no enzimáticas que, si bien se les ha asignado denominaciones diferentes
a las de sus análogas bacterianas, actúan de manera muy similar a ellas.
La replicación del DNA eucariota, sin embargo, presenta una serie de
particularidades que es conveniente poner de manifiesto: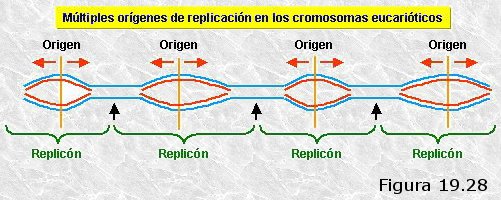
-
Los cromosomas eucariotas presentan múltiples orígenes de replicación que se alternan con las correspondientes secuencias de terminación, de manera que en el proceso replicativo varias burbujas de replicación están activas simultáneamente y van creciendo en ambos sentidos hasta que se funden con sus vecinas. Cada unidad replicativa, incluyendo su origen de replicación y delimitada por las correspondientes secuencias de terminación, recibe el nombre de replicón (Figura 19.28).
-
Las DNA polimerasas eucariotas son sensiblemente más lentas que las bacterianas (algunas decenas de nucleótidos por segundo frente a unos 1.000 nucleótidos por segundo). Tal lentitud se compensa con la existencia de orígenes múltiples de replicación.
-
Los fragmentos de Okazaki son más cortos que los de las células procariotas.
-
La eliminación de los cebadores es realizada por una exonucleasa 5’à3’ que es independiente de la polimerasa. No existe un enzima análogo a la DNA polimerasa I de E. coli. La sustitución de los cebadores por DNA, una vez eliminados éstos, es realizada por las mismas DNA polimerasas responsables de la síntesis de los fragmentos.
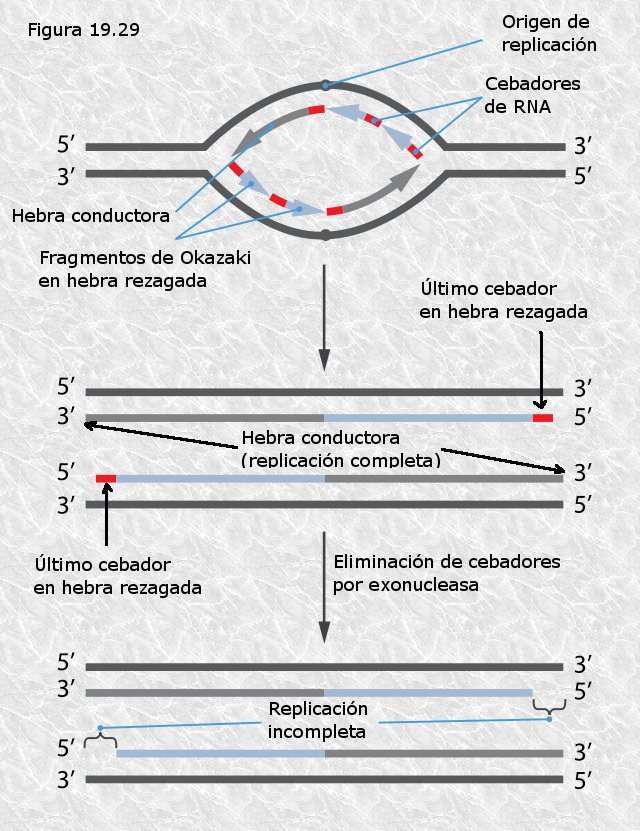 Mención aparte merece el problema del final de la
replicación en los extremos de los cromosomas eucariotas denominados
telómeros. Este problema no se da en las células procariotas por
presentar éstas cromosomas circulares y por lo tanto carentes de
extremos. En los telómeros la replicación de la hebra conductora
finaliza sin complicaciones ya que las DNA polimerasas finalizan la
síntesis en el lugar exacto en el que finaliza el molde. Por el
contrario, en la hebra rezagada el cebador del último fragmento de
Okazaki es colocado exactamente en el extremo 5’, emparejado con la
cadena molde; cuando la exonucleasa 5’à3’
elimina este cebador ninguna DNA polimerasa podrá rellenar el
hueco que deja (Figura
19.29). De este modo, la replicación queda inconclusa en el extremo
5’ y cuando en el siguiente ciclo replicativo la cadena así acortada
sirva de molde para la síntesis de su complementaria se habrá perdido
definitivamente un tramo de la cadena polinucleotídica.
Mención aparte merece el problema del final de la
replicación en los extremos de los cromosomas eucariotas denominados
telómeros. Este problema no se da en las células procariotas por
presentar éstas cromosomas circulares y por lo tanto carentes de
extremos. En los telómeros la replicación de la hebra conductora
finaliza sin complicaciones ya que las DNA polimerasas finalizan la
síntesis en el lugar exacto en el que finaliza el molde. Por el
contrario, en la hebra rezagada el cebador del último fragmento de
Okazaki es colocado exactamente en el extremo 5’, emparejado con la
cadena molde; cuando la exonucleasa 5’à3’
elimina este cebador ninguna DNA polimerasa podrá rellenar el
hueco que deja (Figura
19.29). De este modo, la replicación queda inconclusa en el extremo
5’ y cuando en el siguiente ciclo replicativo la cadena así acortada
sirva de molde para la síntesis de su complementaria se habrá perdido
definitivamente un tramo de la cadena polinucleotídica.
El acortamiento de los telómeros en cada ciclo de división
celular a causa del fenómeno descrito puede suponer la pérdida de
información genética importante y con ello el deterioro y la muerte de
la célula. Para paliar este grave problema los organismos eucariontes se
han dotado de un enzima, la telomerasa, que tiene la función de
reparar los telómeros en cada ciclo de división celular evitando así su
acortamiento. La
telomerasa es un enzima con actividad de DNA polimerasa que
lleva incorporado como grupo prostético un oligonucleótido de RNA que
sirve de molde para sintetizar una cadena complementaria de DNA. Los
telómeros poseen unas secuencias repetitivas de DNA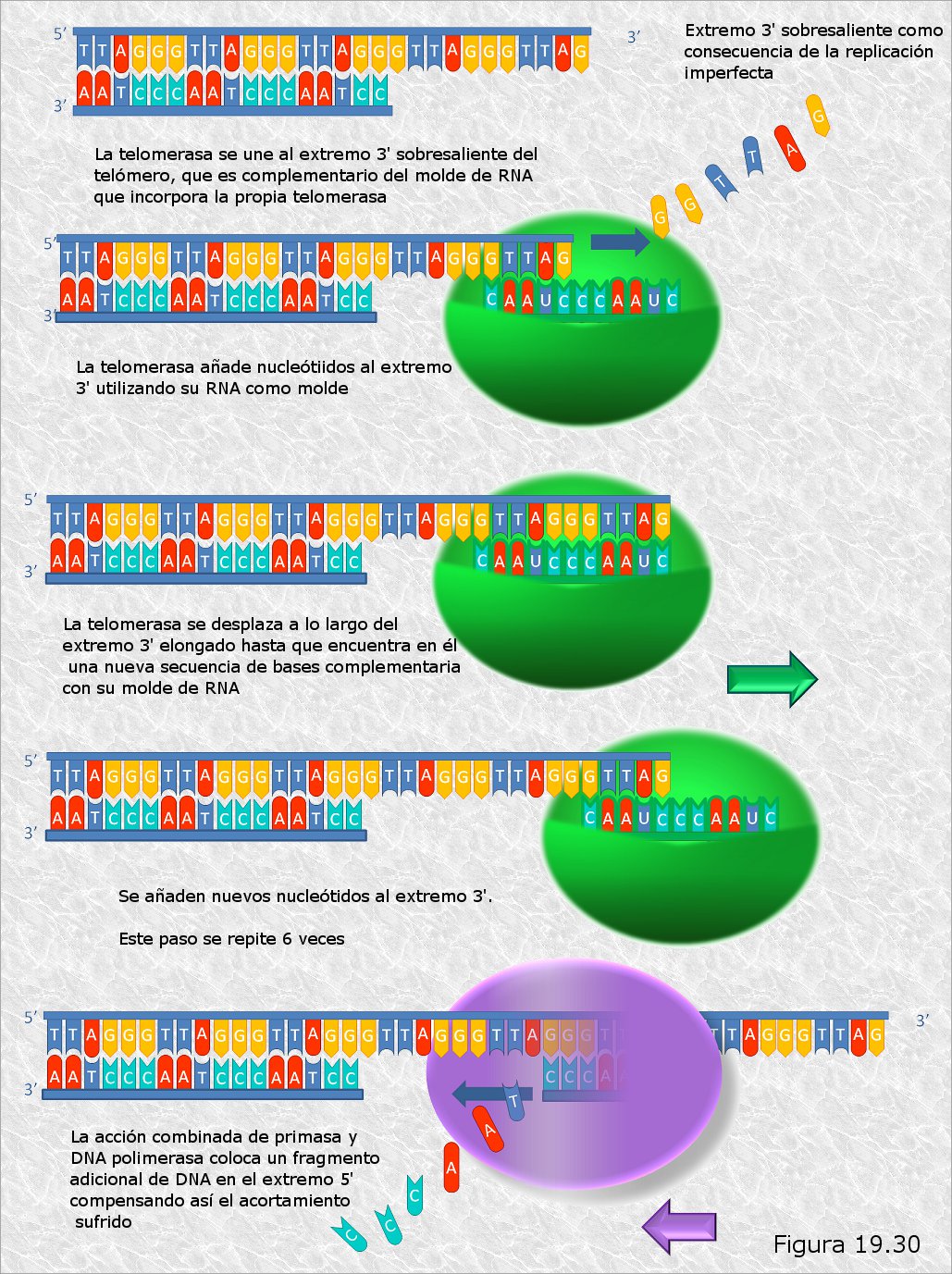 parcialmente
complementarias con el molde de RNA interno de la telomerasa.
Cuando estas secuencias se aparean, dicho molde interno es utilizado
para alargar el extremo 3’ sobresaliente del telómero. Se producen
varios ciclos de alargamiento y a continuación la maquinaria replicativa
ordinaria coloca un fragmento de Okazaki adicional en el extremo 5’
defectuoso, de manera que, aunque posteriormente se eliminará el cebador
de este fragmento, el tramo de DNA así sintetizado compensa el
acortamiento del telómero (Figura
19.30).
parcialmente
complementarias con el molde de RNA interno de la telomerasa.
Cuando estas secuencias se aparean, dicho molde interno es utilizado
para alargar el extremo 3’ sobresaliente del telómero. Se producen
varios ciclos de alargamiento y a continuación la maquinaria replicativa
ordinaria coloca un fragmento de Okazaki adicional en el extremo 5’
defectuoso, de manera que, aunque posteriormente se eliminará el cebador
de este fragmento, el tramo de DNA así sintetizado compensa el
acortamiento del telómero (Figura
19.30).
{kind=link}
Gracias a la acción de la telomerasa la longitud de los telómeros permanece aproximadamente constante a lo largo de las sucesivas generaciones celulares. Sin embargo, se ha comprobado que la actividad de este enzima varía considerablemente entre distintos tipos de células eucariotas. Se muestra particularmente activa en los organismos eucariontes unicelulares y en las células germinales de los pluricelulares (células en las que la conservación de la integridad del genoma es inexcusable), pero esta actividad decae considerablemente en las células somáticas de estos últimos. Así, cuando estas células se dividen sucesivamente sus telómeros se van acortando hasta que su información genética resulta afectada con el consiguiente deterioro y muerte celular.
Se ha especulado mucho con la idea de que el acortamiento de los telómeros en las células somáticas pueda estar relacionado con los procesos de envejecimiento general del organismo y de que una potenciación de la actividad de la telomerasa en estas células pudiera resultar útil para retrasarlo. Existe un grave inconveniente: la actividad de la telomerasa es particularmente alta en la mayoría de las células tumorales. En realidad, la desactivación de este enzima en las células somáticas podría funcionar como un eficaz mecanismo de prevención del cáncer, ya que el acortamiento de los telómeros actuaría como una especie de reloj biológico que conduciría a la muerte celular antes de que una célula pueda acumular las mutaciones necesarias para convertirse en tumoral. Por el contrario, las células portadoras de una mutación que reactive la telomerasa serían candidatas a convertirse en cancerígenas. El bloqueo selectivo de la actividad de la telomerasa en estas células podría constituir una vía para el tratamiento de esta enfermedad.
5.- EXPRESIÓN GÉNICA.
 Una de los interrogantes que ya se planteaban los genetistas
clásicos acerca de la función del gen mendeliano era el cómo dicho gen
era capaz de determinar el carácter hereditario del que era responsable.
En otras palabras: ¿de qué manera el genotipo determina el fenotipo? Los
primeros pasos en la resolución de esta cuestión se dieron mucho antes
de que se elucidara la naturaleza del gen; en realidad casi al mismo
tiempo que se redescubrían los trabajos de Mendel a comienzos del siglo
XX.
Una de los interrogantes que ya se planteaban los genetistas
clásicos acerca de la función del gen mendeliano era el cómo dicho gen
era capaz de determinar el carácter hereditario del que era responsable.
En otras palabras: ¿de qué manera el genotipo determina el fenotipo? Los
primeros pasos en la resolución de esta cuestión se dieron mucho antes
de que se elucidara la naturaleza del gen; en realidad casi al mismo
tiempo que se redescubrían los trabajos de Mendel a comienzos del siglo
XX.
En 1902 el médico inglés Archibald Garrod (Figura 19.31) estudió los antecedentes familiares de algunos de sus pacientes de alcaptonuria (afección artrítica cuyo principal síntoma es la excreción de orina de color vino) y llegó a la conclusión de que se trataba de una enfermedad hereditaria. Garrod dedujo asimismo que la alcaptonuria consistía en una alteración del metabolismo del nitrógeno que daba lugar a la excreción de una sustancia de color oscuro en lugar de la urea, que es el componente normal de la orina. Su hipótesis, que resultó ser acertada, era que estos pacientes eran homocigotos para el alelo recesivo de un gen que controla una reacción metabólica enzimáticamente catalizada. Este alelo determinaba la aparición de un fallo en dicha reacción metabólica de modo que se producía la acumulación de la sustancia que en condiciones normales sería transformada en la reacción.
Garrod acuñó en 1908 la frase “errores congénitos del metabolismo” para referirse a las alteraciones de los genes que controlaban las reacciones metabólicas enzimáticamente catalizadas, siendo la suya la primera referencia a la relación entre genes y enzimas. Unos treinta años más tarde G. W. Beadle y E.L. Tatum realizaron una serie experimentos que pusieron de manifiesto esta relación.
 Beadle y Tatum (Figura
19.32) utilizaron para sus experiencias el hongo Neurospora
crassa (moho del pan), que por su facilidad de cultivo y
manipulación en el laboratorio se había revelado como una gran
herramienta en la investigación genética. En primer lugar aislaron una
serie de mutantes del hongo incapaces de crecer en un medio de cultivo
mínimo (medio con las sustancias imprescindibles para permitir el
crecimiento del moho) en el que sí crecían los ejemplares salvajes.
Estos mutantes, denominados mutantes auxotrofos debían su
incapacidad a un fallo en su metabolismo que les impedía sintetizar
algún metabolito concreto (normalmente alguno de los veinte aminoácidos
o alguna vitamina). Beadle y Tatum identificaron a continuación cual era
la sustancia que necesitaba cada mutante suplementando diferentes
muestras del medio de cultivo mínimo con una amplia variedad de
metabolitos candidatos. El medio suplementado con la sustancia buscada
permitía el crecimiento del mutante en cuestión. Perseverando con esta
estrategia Beadle y Tatum consiguieron identificar no sólo el metabolito
imprescindible para cada mutante, sino la reacción metabólica
enzimáticamente catalizada que se encontraba bloqueada en la ruta que
conducía a su síntesis.
Beadle y Tatum (Figura
19.32) utilizaron para sus experiencias el hongo Neurospora
crassa (moho del pan), que por su facilidad de cultivo y
manipulación en el laboratorio se había revelado como una gran
herramienta en la investigación genética. En primer lugar aislaron una
serie de mutantes del hongo incapaces de crecer en un medio de cultivo
mínimo (medio con las sustancias imprescindibles para permitir el
crecimiento del moho) en el que sí crecían los ejemplares salvajes.
Estos mutantes, denominados mutantes auxotrofos debían su
incapacidad a un fallo en su metabolismo que les impedía sintetizar
algún metabolito concreto (normalmente alguno de los veinte aminoácidos
o alguna vitamina). Beadle y Tatum identificaron a continuación cual era
la sustancia que necesitaba cada mutante suplementando diferentes
muestras del medio de cultivo mínimo con una amplia variedad de
metabolitos candidatos. El medio suplementado con la sustancia buscada
permitía el crecimiento del mutante en cuestión. Perseverando con esta
estrategia Beadle y Tatum consiguieron identificar no sólo el metabolito
imprescindible para cada mutante, sino la reacción metabólica
enzimáticamente catalizada que se encontraba bloqueada en la ruta que
conducía a su síntesis.
Los resultados obtenidos condujeron a Beadle y Tatum a la formulación de la teoría “un gen – un enzima”, que viene a afirmar que la función primaria de un gen consiste en dirigir la formación de un (y sólo un) enzima. Esta teoría representa el primer avance en el conocimiento del complejo camino que conduce desde el genotipo al fenotipo. A partir de su formulación el concepto de gen dejó de depender de la existencia de dos o más variantes alélicas que permitiesen detectar su existencia para convertirse en algo mucho más “tangible”, aunque todavía se ignorase su naturaleza físico-química. La función de los genes parecía ser la de dirigir el metabolismo celular a través del control de la síntesis de los enzimas responsables del mismo.
A medida que avanzaba el conocimiento de la estructura de las proteínas y se establecía que era la secuencia de aminoácidos la que determinaba la conformación tridimensional de estas macromoléculas y, a través de ella, su función biológica, surgió la idea de que la función del gen no podía ser otra que la de dirigir en ensamblaje ordenado de los distintos aminoácidos durante el proceso de síntesis de la proteína correspondiente. Dicho de otro modo, la información genética almacenada en el gen se expresaba en forma de secuencia de aminoácidos de una proteína, que a su vez especificaba su conformación tridimensional y con ello su función biológica. Además, el efecto de las mutaciones consistiría en producir alteraciones en la secuencia de aminoácidos que darían lugar a conformaciones tridimensionales alteradas afectando así a la función biológica de la proteína. Diversos experimentos realizados a finales de la década de años 40 del siglo XX pusieron de manifiesto la veracidad de estas aseveraciones. En particular, el descubrimiento de que la anemia falciforme (una enfermedad hereditaria frecuente en algunas poblaciones africanas) se debía a la sustitución de un solo aminoácido en una de las cadenas polipeptídicas de la hemoglobina de los pacientes afectados, constituyó la primera prueba directa acerca de la relación entre genes y proteínas en el ser humano.
En su versión actual, a la vista de que muchos enzimas y proteínas no enzimáticas están formadas por varias subunidades, la teoría “un gen – un enzima” se ha reformulado como teoría “un gen – una cadena polipeptídica”. Es necesario resaltar que esta teoría pertenece al campo de lo que se ha dado en llamar genética clásica: el gen, que según la teoría informa para la síntesis de un enzima sigue siendo una entidad casi tan abstracta como el gen mendeliano. La teoría no proporciona respuestas a la pregunta ¿de qué manera el gen controla la síntesis de un enzima? Estas repuestas habrían de venir de la mano de una nueva área del conocimiento: la genética molecular.
5.1.- EL “DOGMA CENTRAL”.
El descubrimiento de la naturaleza del material genético arrojó nueva luz sobre la relación entre genes y proteínas. En efecto, si la información genética se encontraba cifrada en forma de la secuencia de nucleótidos del DNA, resultaba claro, a la luz de la teoría “un gen – un enzima”, que la expresión de esta información debería consistir en una “traducción” de la secuencia de nucleótidos de un gen a la secuencia de aminoácidos de una proteína. Ahora bien, ¿cómo se lleva a cabo este proceso?
Como ya se ha comentado con anterioridad, los primeros estudios acerca del RNA sugerían que este ácido nucleico guardaba cierta relación con la síntesis de proteínas y que pronto se estableció que unas partículas citoplasmáticas ricas en RNA, los ribosomas, eran el lugar en donde tenía lugar dicha síntesis. Resultaba evidente que el DNA, en su sede nuclear, no podía presidir directamente el ensamblaje de los aminoácidos que tenía lugar en el citoplasma. Así surgió la idea de que la expresión génica era un proceso en dos etapas: la primera, que tendría lugar en el núcleo, sería una transcripción de la información genética almacenada en el DNA a un intermediario de RNA; la segunda, que tendría lugar en los ribosomas, sería una traducción de dicha información a la secuencia de aminoácidos de una proteína. No está claro quien fue el primer investigador en formular estas ideas, aunque parece claro que formaban parte del pensamiento de Watson y Crick muy poco después del descubrimiento de la doble hélice.
El primer intento de explicación para el mecanismo de la expresión génica postulaba que el DNA se transcribía dando lugar al componente de RNA de los ribosomas, de manera que cada gen producía así un ribosoma específico que se encargaba de sintetizar una proteína (y sólo una). Tal mecanismo, conocido como la hipótesis “un gen – un ribosoma – una proteína”, tuvo que ser muy pronto revisado a la luz de los experimentos realizados al efecto. Cuando un bacteriófago infecta una célula bacteriana la maquinaria bioquímica de ésta es inmediatamente “secuestrada” por el virus y se desvía hacia la síntesis de las proteínas de la progenie viral. Es de esperar, por lo tanto, que, de ser correcta la hipótesis “un gen – un ribosoma – una proteína”, la infección vírica dispare la producción de nuevos ribosomas que se encargarán de sintetizar las proteínas del virus. Los experimentos llevados a cabo utilizando el marcaje con isótopos radiactivos en células de E. coli infectadas por el bacteriófago T4 indicaban que, por el contrario, la infección detenía la producción de nuevos ribosomas y que, por lo tanto, las proteínas de la progenie viral eran sintetizadas por ribosomas ya presentes en la célula antes de la infección. Además, el único tipo de RNA que aparecía en las bacterias infectadas no era el componente habitual de los ribosomas, sino un nuevo tipo de RNA muy inestable y que se encontraba débilmente asociado a éstos.
 Estos resultados experimentales condujeron a F. Jacob y J. Monod (Figura
19.33) a formular en 1961 un mecanismo alternativo para el proceso
de expresión de la información genética. El RNA ribosómico no es el
patrón directo que dirige el ensamblaje de los aminoácidos y, por lo
tanto, los ribosomas no están especializados en la síntesis de
determinadas cadenas polipeptídicas. En lugar de ello, la secuencia de
nucleótidos del DNA se transcribe a moléculas de un tipo diferente de
RNA, el RNA mensajero, que emigran posteriormente al citoplasma
para allí entrar temporalmente en combinación con los ribosomas. El
complejo formado por los ribosomas y el RNA mensajero sería el encargado
de dirigir el ensamblaje de los aminoácidos de una cadena polipeptídica,
siendo el ribosoma el lugar de ensamblaje y el RNA mensajero la
“plantilla” que contiene la información necesaria para hacerlo. El
mRNA se degrada rápidamente después de haber dirigido la síntesis de
unas pocas moléculas de proteína, lo que explicaría la gran
inestabilidad que presentaba en los experimentos.
Estos resultados experimentales condujeron a F. Jacob y J. Monod (Figura
19.33) a formular en 1961 un mecanismo alternativo para el proceso
de expresión de la información genética. El RNA ribosómico no es el
patrón directo que dirige el ensamblaje de los aminoácidos y, por lo
tanto, los ribosomas no están especializados en la síntesis de
determinadas cadenas polipeptídicas. En lugar de ello, la secuencia de
nucleótidos del DNA se transcribe a moléculas de un tipo diferente de
RNA, el RNA mensajero, que emigran posteriormente al citoplasma
para allí entrar temporalmente en combinación con los ribosomas. El
complejo formado por los ribosomas y el RNA mensajero sería el encargado
de dirigir el ensamblaje de los aminoácidos de una cadena polipeptídica,
siendo el ribosoma el lugar de ensamblaje y el RNA mensajero la
“plantilla” que contiene la información necesaria para hacerlo. El
mRNA se degrada rápidamente después de haber dirigido la síntesis de
unas pocas moléculas de proteína, lo que explicaría la gran
inestabilidad que presentaba en los experimentos.
Los puntos de vista de Jacob y Monod fueron confirmados en experimentos de hibridación DNA-RNA. El RNA inestable que aparecía en el citoplasma bacteriano tras la infección fágica fue extraído y se comprobó que hibridaba con el DNA del virus una vez desnaturalizado. Ello demostraba que la secuencia de nucleótidos de RNA era ciertamente complementaria con la correspondiente del DNA vírico, que es lo que el modelo de Jacob y Monod predecía para la molécula que habían denominado RNA mensajero. Hay que resaltar que en los híbridos DNA-RNA que se obtenían en estos experimentos los pares de bases A-T se ven sustituidos por pares A-U, que forman igualmente dos puentes de hidrógeno entre sí. Experimentos posteriores del mismo tenor arrojaron resultados similares para el mRNA bacteriano en relación con los genes bacterianos. Por otra parte, también se comprobó que no sólo el mRNA sino todos los tipos de RNA se sintetizan a partir de moldes de DNA mediante un proceso de transcripción.
Las ideas hasta aquí expuestas acerca del mecanismo de la expresión génica constituyen el núcleo de lo que se ha dado en llamar “el dogma central de la biología molecular”, que viene a afirmar que la información genética fluye unidireccionalmente desde el DNA hacia las proteínas a través de un intermediario de RNA. El “dogma”, así formulado por Francis Crick, hubo de ser revisado a consecuencia del descubrimiento de enzimas capaces de sintetizar DNA a partir de moldes de DNA, en un proceso que se denominó transcripción inversa (Figura 19.34).
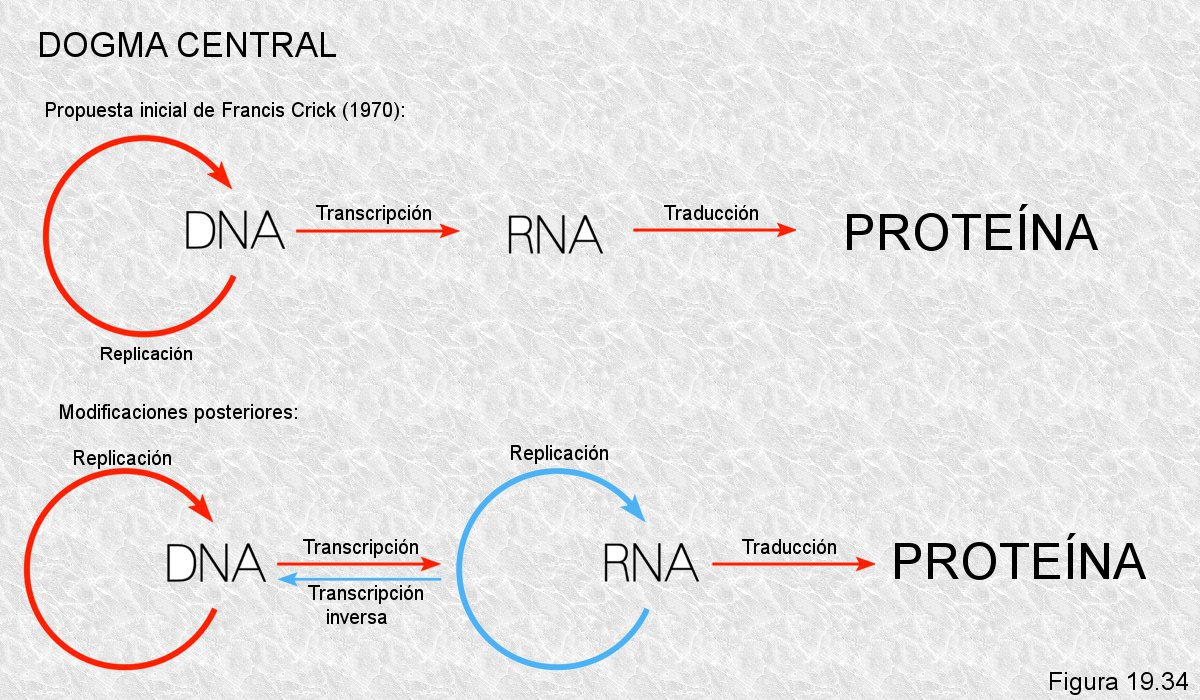
5.2.- TRANSCRIPCIÓN.
Los aspectos químicos de la síntesis del RNA son muy similares a los de la replicación del DNA. La adición de nucleótidos a una cadena de RNA en crecimiento necesita también de nucleótidos trifosfato que se incorporan según la siguiente reacción:
RNA (n) + NTP à RNA (n+1) + PPi
 La reacción es catalizada por el enzima RNA polimerasa
(Figura 19.35),
descubierto en 1960 por S. Weiss en extractos de E. coli. Desde
entonces se han encontrado RNA polimerasas en todo tipo de
células, compartiendo todas ellas una serie de características. Las
RNA polimerasas necesitan un molde de DNA sin el cual son incapaces
de polimerizar nucleótidos; a diferencia de las DNA polimerasas,
pueden iniciar cadenas polinucleotídicas colocando un primer nucleótido
y añadiendo a éste los siguientes; todas llevan a cabo la síntesis en
dirección 5’à3’ recorriendo
el molde en la dirección opuesta. Las RNA polimerasas son enzimas
multiméricos formados por varias subunidades entre las que destaca la
llamada factor σ, responsable de reconocer el lugar de inicio de
la transcripción.
La reacción es catalizada por el enzima RNA polimerasa
(Figura 19.35),
descubierto en 1960 por S. Weiss en extractos de E. coli. Desde
entonces se han encontrado RNA polimerasas en todo tipo de
células, compartiendo todas ellas una serie de características. Las
RNA polimerasas necesitan un molde de DNA sin el cual son incapaces
de polimerizar nucleótidos; a diferencia de las DNA polimerasas,
pueden iniciar cadenas polinucleotídicas colocando un primer nucleótido
y añadiendo a éste los siguientes; todas llevan a cabo la síntesis en
dirección 5’à3’ recorriendo
el molde en la dirección opuesta. Las RNA polimerasas son enzimas
multiméricos formados por varias subunidades entre las que destaca la
llamada factor σ, responsable de reconocer el lugar de inicio de
la transcripción.
Otro rasgo importante del proceso de transcripción es su asimetría. En una secuencia dada de DNA sólo una de las cadenas polinucleotídicas, la llamada hebra codificadora o hebra con sentido, se transcribe a RNA; la otra cadena, llamada hebra estabilizadora o hebra sin sentido no contiene información para la síntesis de proteínas, aunque es imprescindible para, en el proceso de replicación, servir de molde para la síntesis de otra hebra codificadora. En general, en las células procariotas la hebra codificadora es la misma en toda la extensión del cromosoma. Por el contrario, en las células eucariotas la hebra codificadora cambia de unos genes a otros.
En el proceso de transcripción podemos distinguir tres fases: iniciación, elongación y terminación:
FASE DE INICIACIÓN.
La transcripción se inicia para cada gen o grupo de genes en una secuencia específica de DNA denominada promotor (Figura 19.36). Se han identificado varias secuencias promotoras, siendo su característica común más apreciable su riqueza en pares A-T. La RNA polimerasa con su factor σ incorporado reconoce al promotor y provoca en él el desenrollamiento local de la doble hélice y la consiguiente apertura de una burbuja de transcripción. A continuación coloca el primer nucleótido a una distancia de 10 pares de bases del final del promotor. Una vez iniciada la síntesis, el factor σ ya no es necesario y se desprende (Figura 19.35).
FASE DE ELONGACIÓN.
La RNA polimerasa va añadiendo nucleótidos en dirección 5’à3’. El mismo enzima presenta una actividad helicasa que va abriendo la burbuja de transcripción a medida que avanza. La colocación de los sucesivos nucleótidos se atiene a las reglas de apareamiento de bases nitrogenadas, con la salvedad conocida de que el par A-T es sustituido por A-U. De manera similar a lo que ocurre en la replicación del DNA, la cadena en crecimiento forma una doble hélice con la cadena molde (Figura 19.36). Sin embargo, a diferencia de la replicación, este dúplex es transitorio: la cadena de RNA se va desprendiendo de su molde de DNA a medida que se va sintetizando; en un instante dado durante la transcripción el dúplex híbrido DNA-RNA apenas ocupa una vuelta de hélice (entre ocho y doce nucleótidos). A medida que se va desprendiendo el RNA recién sintetizado la burbuja de transcripción se va cerrando tras la RNA polimerasa.
FASE DE TERMINACIÓN.
La RNA polimerasa reconoce determinadas secuencias de nucleótidos en el DNA, llamadas terminadores, que constituyen una señal para la interrupción de la síntesis de RNA y el desprendimiento del enzima con el consiguiente cierre de la burbuja de transcripción. En muchos casos los terminadores son secuencias palindrómicas (se leen igual en uno y otro sentido) que propician la formación de un bucle interno en el RNA. La formación de este bucle induce el desprendimiento del enzima.
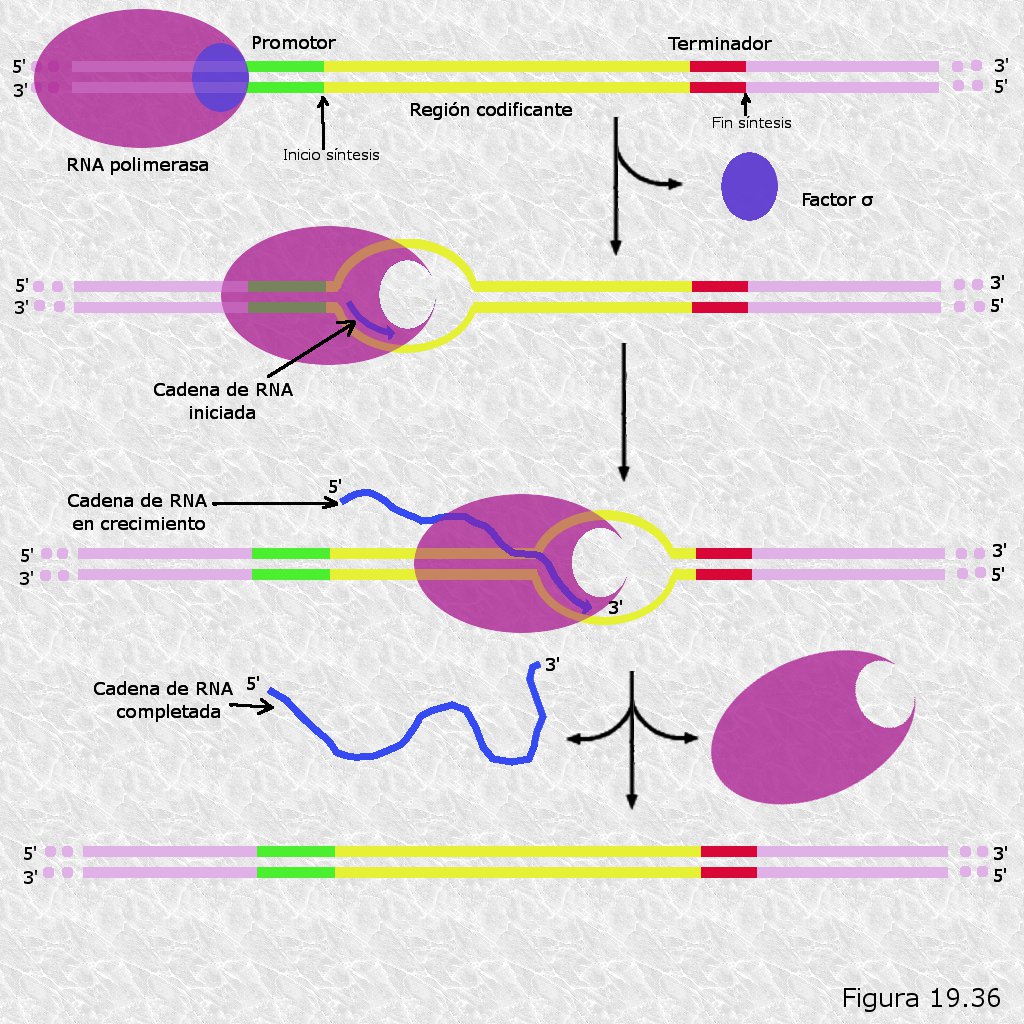
Los procesos de transcripción hasta aquí descritos son básicamente iguales en las células procariotas y eucariotas. Sin embargo, existen algunas diferencias entre ambos tipos celulares en lo que se refiere al destino inmediato de los RNAs producto de la transcripción.
En las células procariotas los mRNA sintetizados son directamente utilizados en los ribosomas para la síntesis de proteínas sin necesidad de ninguna transformación previa. Los mRNA procariotas son policistrónicos, es decir, una sola molécula de RNA es el resultado de la transcripción de varios genes contiguos y su traducción por los ribosomas origina varias proteínas diferentes. Asimismo, las secuencias que codifican los tRNA y las que codifican los rRNA son transcritas en una sola molécula, el transcrito primario, que después es cortada por la acción de nucleasas específicas, dando lugar a las correspondientes moléculas de RNA biológicamente activas. Por otra parte, los procesos de transcripción y traducción en las células procariotas no están separados en el tiempo: la traducción comienza por el extremo 5’ antes de que la RNA polimerasa haya finalizado la síntesis del mensajero.
En las células eucariotas los procesos de transcripción y traducción están necesariamente separados en el tiempo, ya que el primero ocurre en el núcleo y el segundo en el citoplasma celular, debiendo los RNA sintetizados atravesar los poros nucleares para incorporarse a sus respectivas misiones en la síntesis de proteínas. Los productos de la transcripción en las células eucariotas sufren una serie de complejos procesos de maduración que pasaremos a discutir a continuación.
5.3.- GENES FRAGMENTADOS Y MADURACIÓN DEL RNA.
 Los estudios realizados acerca del mecanismo de expresión de
la información genética durante las décadas de los años 60 y 70 del
siglo XX indicaban que para cada gen una secuencia continua de
nucleótidos del DNA codificaba la correspondiente secuencia de
aminoácidos de la proteína correspondiente. En 1977 P.A. Sharp y R. J.
Roberts (Figura
19.37) obtuvieron unos resultados experimentales desconcertantes que
venían a poner en cuestión tales ideas. En determinadas condiciones
experimentales es posible obtener híbridos estables entre mRNAs y
las secuencias de DNA que los codifican. Sharp y Roberts obtuvieron este
tipo de híbridos con ácidos nucleicos de un adenovirus y los
observaron al microscopio electrónico, comprobando que el mRNA y el DNA
codificante no hibridaban en toda su longitud, sino que aparecían lazos
formados por tramos de DNA de cadena sencilla. Ello demostraba que había
segmentos del DNA que no estaban representados en el correspondiente
mRNA y que por lo tanto los genes del virus estaban fragmentados en
tramos codificantes, a los que se denominó exones y tramos no
codificantes, a los que se denominó intrones.
Los estudios realizados acerca del mecanismo de expresión de
la información genética durante las décadas de los años 60 y 70 del
siglo XX indicaban que para cada gen una secuencia continua de
nucleótidos del DNA codificaba la correspondiente secuencia de
aminoácidos de la proteína correspondiente. En 1977 P.A. Sharp y R. J.
Roberts (Figura
19.37) obtuvieron unos resultados experimentales desconcertantes que
venían a poner en cuestión tales ideas. En determinadas condiciones
experimentales es posible obtener híbridos estables entre mRNAs y
las secuencias de DNA que los codifican. Sharp y Roberts obtuvieron este
tipo de híbridos con ácidos nucleicos de un adenovirus y los
observaron al microscopio electrónico, comprobando que el mRNA y el DNA
codificante no hibridaban en toda su longitud, sino que aparecían lazos
formados por tramos de DNA de cadena sencilla. Ello demostraba que había
segmentos del DNA que no estaban representados en el correspondiente
mRNA y que por lo tanto los genes del virus estaban fragmentados en
tramos codificantes, a los que se denominó exones y tramos no
codificantes, a los que se denominó intrones.

En los años que siguieron se realizaron experimentos
similares con otros organismos y se pudo comprobar que los genes
fragmentados están ampliamente difundidos en la naturaleza. Por ejemplo,
el gen que codifica la ovoalbúmina de gallina, uno de los primeros en
ser analizado respecto a su fragmentación, presenta 7 intrones a
lo largo de sus 7700 pares de bases, con longitudes que van desde los
251 a los 1.600 pares de bases (Figura
19.38).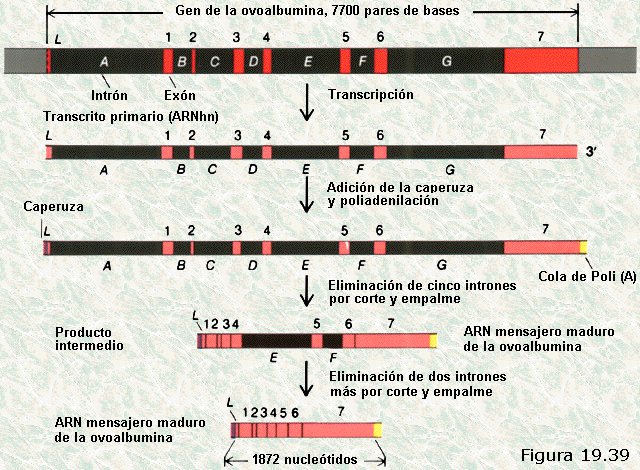
Los genes fragmentados abundan mucho más entre los organismos eucariontes que entre los procariontes. En los vertebrados la inmensa mayoría del genoma está salpicada de intrones de longitud y distribución muy variables. Sólo unos pocos genes de estos organismos (los que codifican proteínas histónicas) carecen de ellos. En el extremo opuesto se encuentran algunas levaduras como Saccharomyces cerevisiae en las que los intrones apenas existen. En el mundo procariota los genes fragmentados son muy escasos, estando su presencia prácticamente limitada a un grupo reducido denominado arqueobacterias. También se han detectado intrones en algunos grupos de virus.
La correcta expresión de la información de los genes fragmentados requiere la eliminación de las secuencias no codificantes en algún momento del proceso. Esta eliminación se lleva a cabo sobre las moléculas de RNA recién sintetizadas, los transcritos primarios, en un proceso denominado corte y empalme (splicing en inglés).
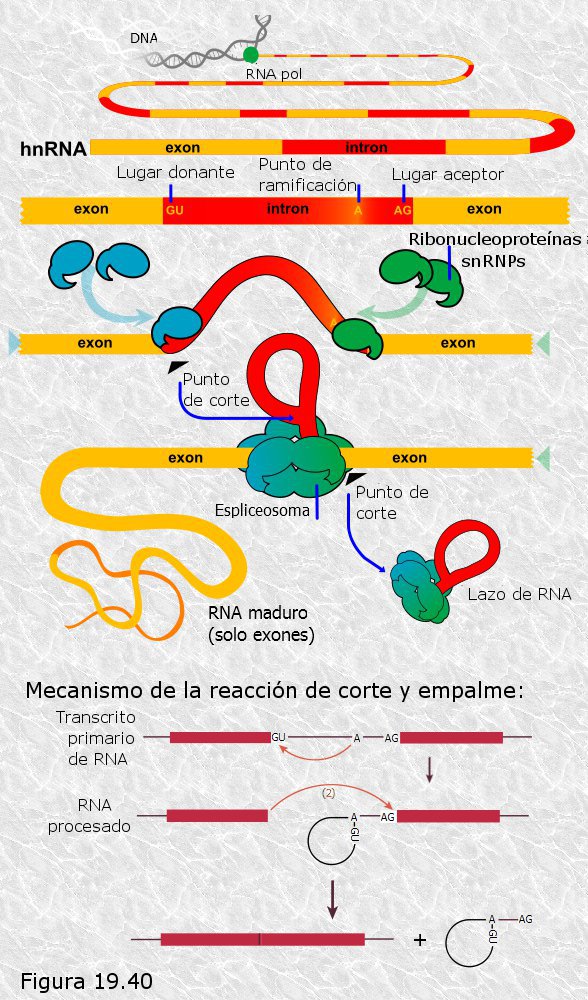 Existen varios tipos de intrones en relación con el
mecanismo de corte y empalme que actúa sobre ellos. El grupo más
numeroso es el que incluye a los intrones presentes en los
transcritos primarios precursores de los mRNA eucariotas. En ellos el
mecanismo de corte y empalme requiere la participación de unas
ribonucleoproteínas con actividad enzimática de la clase denominada
ribonucleoproteína nuclear pequeña (snRNP). Estas proteínas llevan
incorporado como grupo prostético una molécula de RNA del tipo
denominado RNA nuclear pequeño (snRNA), estando presentes en el
núcleo de las células eucariotas. Varias de estas ribonucleoproteínas se
agrupan para formar un complejo denominado espliceosoma, que es
el encargado del proceso de corte y empalme. Los espliceosomas
catalizan una reacción química consistente en el intercambio de un
enlace fosfodiéster por otro con la consiguiente eliminación del
intrón y el enlace de los dos exones situados a ambos lados
de éste (Figura
19.40). Por medio de un apareamiento específico de bases con su
componente snRNA los espliceosomas reconocen secuencias
específicas del transcrito primario que señalan el comienzo y final de
los distintos intrones. El corte y empalme debe realizarse con
una gran precisión, ya que cualquier error, aunque sólo afecte a un
único nucleótido, se traduciría en un error que afectaría a toda la
cadena polipeptídica codificada.
Existen varios tipos de intrones en relación con el
mecanismo de corte y empalme que actúa sobre ellos. El grupo más
numeroso es el que incluye a los intrones presentes en los
transcritos primarios precursores de los mRNA eucariotas. En ellos el
mecanismo de corte y empalme requiere la participación de unas
ribonucleoproteínas con actividad enzimática de la clase denominada
ribonucleoproteína nuclear pequeña (snRNP). Estas proteínas llevan
incorporado como grupo prostético una molécula de RNA del tipo
denominado RNA nuclear pequeño (snRNA), estando presentes en el
núcleo de las células eucariotas. Varias de estas ribonucleoproteínas se
agrupan para formar un complejo denominado espliceosoma, que es
el encargado del proceso de corte y empalme. Los espliceosomas
catalizan una reacción química consistente en el intercambio de un
enlace fosfodiéster por otro con la consiguiente eliminación del
intrón y el enlace de los dos exones situados a ambos lados
de éste (Figura
19.40). Por medio de un apareamiento específico de bases con su
componente snRNA los espliceosomas reconocen secuencias
específicas del transcrito primario que señalan el comienzo y final de
los distintos intrones. El corte y empalme debe realizarse con
una gran precisión, ya que cualquier error, aunque sólo afecte a un
único nucleótido, se traduciría en un error que afectaría a toda la
cadena polipeptídica codificada.
La maduración de los transcritos primarios precursores de
los mRNAs eucariotas incluye, además de la eliminación de los
intrones, otros procesos destinados a proporcionar una mayor
estabilidad química a la molécula resultante y evitar que sea
rápidamente degradada por las nucleasas celulares. Uno de ellos
es la adición en el extremo 5’ de una “caperuza” consistente en un
nucleótido cuya base nitrogenada es la 7-metil-guanina. Otro es la
adición en el extremo de una cola de poli-A (una secuencia repetitiva de
entre 20 y 250 nucleótidos cuya base es la adenina). El poli-A se añade
a partir de una posición concreta del transcrito primario después de
haber eliminado una secuencia no codificante del extremo 3’ por acción
de una nucleasa (Figura
19.39). La adición la realiza un enzima denominado poliadenilato
polimerasa, que no necesita molde para sintetizar el poliadenilato.
Existen, además del tipo descrito, otros tres tipos de intrones presentes en moléculas de rRNA, tRNA y precursores de mensajeros de mitocondrias y cloroplastos que no necesitan de la concurrencia de ribonucleoproteínas específicas para ser eliminados. Los intentos de identificar los enzimas responsables del proceso de corte y empalme en estos intrones produjeron resultados sorprendentes cuando en 1982 T. Cech (Figura 19.41) encontró que eran las propias moléculas de RNA las que catalizaban la eliminación de sus propios intrones, en un proceso conocido como auto-splicing. El descubrimiento de moléculas de RNA con actividad catalítica supuso un duro golpe al dogma de la naturaleza proteica de los enzimas y sirvió para alumbrar nuevos puntos de vista acerca del origen de la vida sobre la Tierra que cristalizaron en la hipótesis del “mundo de RNA”.
Se han descrito unos cuantos casos de procesos de maduración alternativos en los que un único transcrito primario puede dar lugar a dos o más mRNA diferentes y, en consecuencia, a polipéptidos diferentes. En tales casos es el número de exones que se incorporan al mensajero y la ordenación de los mismos lo que determina qué proteína se sintetizará finalmente. Un ejemplo de ello lo constituye un transcrito primario que si se procesa en la glándula tiroides da lugar a la hormona calcitonina y si se procesa en el hipotálamo da lugar al péptido CGRP (Figura 19.42).
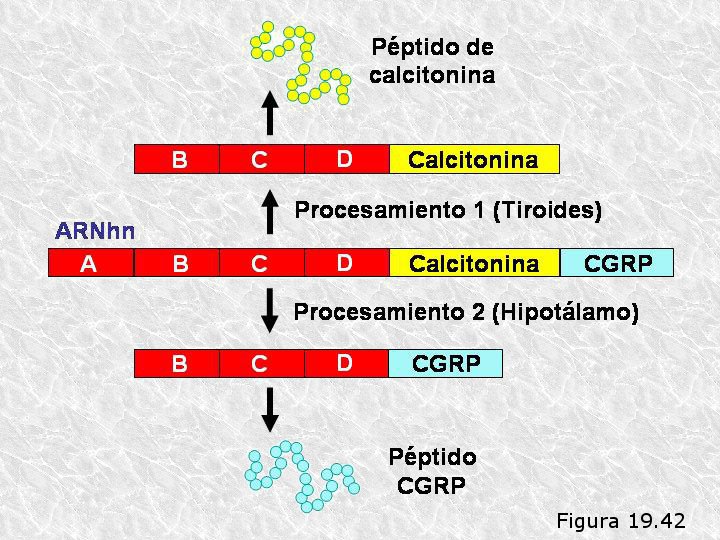
5.4.- EL CÓDIGO GENÉTICO.
Muy poco después de que Watson y Crick propusieran su modelo para la estructura del DNA, asentado ya el convencimiento de que este ácido nucleico era ciertamente la base química de la información genética, muchos investigadores comenzaron a plantearse el problema del código genético. La nueva visión de la teoría “un gen – un enzima”, reformulada a la luz de los nuevos descubrimientos, incorporaba la idea de que la secuencia de aminoácidos del DNA contiene información que especifica la secuencia de aminoácidos de las proteínas. Debía existir, por lo tanto, una clave que relacionase ambos tipos de secuencia, un código que, a modo de diccionario, permitiese a las células vivas traducir la información escrita en el “idioma de los nucleótidos” al “idioma de los aminoácidos”.
Las primeras formulaciones generales acerca del código genético surgieron del simposio anual de Cold Spring Harbor celebrado en el verano de 1953, al que asistieron entre otros Watson y Crick. Resultaba evidente que el código no podía consistir en una correspondencia biunívoca entre bases nitrogenadas y aminoácidos, ya que en el DNA sólo hay cuatro bases mientras que hay veinte aminoácidos en las proteínas. Las “palabras” del código debían, pues, estar formadas por grupos de “letras” representando cada una a una base nitrogenada. Un sencillo cálculo combinatorio eliminaba la posibilidad de que se tratase de grupos de dos bases, pues de este modo sólo se podrían formar 42 = 16 grupos que seguían siendo insuficientes para codificar los veinte aminoácidos. El mismo cálculo realizado para grupos de tres bases indicaba que se podrían formar 43 = 64 grupos diferentes. Así pues, a la vista de que tres era el número mínimo de letras por palabra que permitía codificar la totalidad de los aminoácidos, se llegó a la conclusión de que el código genético constaba de tripletes de bases que especificaban los distintos aminoácidos. La posibilidad de que se tratase de grupos de cuatro o más bases se descartó pues introducía una complejidad innecesaria y ajena a la lógica molecular de las células vivas. Estas conclusiones, obtenidas sobre la base de razonamientos teóricos, fueron confirmadas experimentalmente algunos años después por Francis Crick y Sydney Brenner.
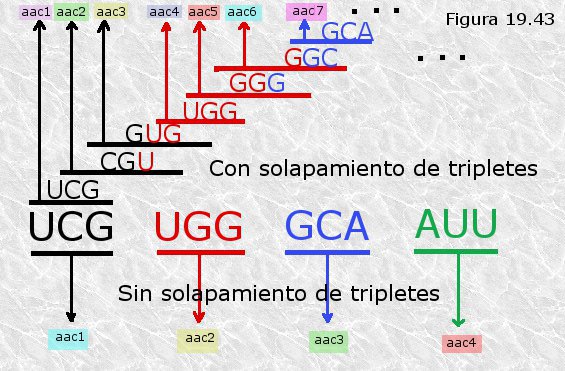 Un primer intento de elaboración de un esquema del código
genético, asumiendo que constaba de tripletes de bases nitrogenadas, fue
acometido por el físico cosmólogo G. Gamow, quien propuso en 1954 que
los diferentes tripletes de bases generaban en la superficie de la
molécula de DNA unas cavidades en las que encajarían las cadenas
laterales de los distintos aminoácidos, proporcionándoles así un molde
sobre el que alinearse antes del ensamblaje. Tal esquema no parecía muy
plausible y además pronto se acumularon pruebas de que el DNA no dirigía
directamente el ensamblaje de los aminoácidos, sino a través de un
intermediario de RNA. Gamow volvió a abordar el problema y elaboró un
nuevo esquema en el que, prescindiendo del mecanismo responsable de la
traducción, proponía un código que contenía tripletes sinónimos (varios
tripletes codifican el mismo aminoácido) que además se encontraban
solapados (cada triplete interviene en la codificación de tres
aminoácidos sucesivos) (Figura
19.43). El código de Gamow presentaba la ventaja de la economía:
eran necesarios menos nucleótidos para codificar el mismo número de
aminoácidos que en uno con tripletes no solapados. Sin embargo,
presentaba un serio inconveniente: la falta de flexibilidad. Un código
con tripletes solapados conllevaría severas restricciones sobre las
secuencias de aminoácidos posibles. Por ejemplo (Figura 19.43), al
aminoácido codificado por el triplete AUU sólo podría seguirle uno
codificado por un triplete UUX (siendo X cualquier base nitrogenada).
Estudios sobre la secuencia de aminoácidos de algunas proteínas
realizados por S. Brenner en 1957 pusieron de manifiesto que en la
naturaleza no existían tales restricciones, por lo que el esquema de
Gamow hubo de ser definitivamente descartado.
Un primer intento de elaboración de un esquema del código
genético, asumiendo que constaba de tripletes de bases nitrogenadas, fue
acometido por el físico cosmólogo G. Gamow, quien propuso en 1954 que
los diferentes tripletes de bases generaban en la superficie de la
molécula de DNA unas cavidades en las que encajarían las cadenas
laterales de los distintos aminoácidos, proporcionándoles así un molde
sobre el que alinearse antes del ensamblaje. Tal esquema no parecía muy
plausible y además pronto se acumularon pruebas de que el DNA no dirigía
directamente el ensamblaje de los aminoácidos, sino a través de un
intermediario de RNA. Gamow volvió a abordar el problema y elaboró un
nuevo esquema en el que, prescindiendo del mecanismo responsable de la
traducción, proponía un código que contenía tripletes sinónimos (varios
tripletes codifican el mismo aminoácido) que además se encontraban
solapados (cada triplete interviene en la codificación de tres
aminoácidos sucesivos) (Figura
19.43). El código de Gamow presentaba la ventaja de la economía:
eran necesarios menos nucleótidos para codificar el mismo número de
aminoácidos que en uno con tripletes no solapados. Sin embargo,
presentaba un serio inconveniente: la falta de flexibilidad. Un código
con tripletes solapados conllevaría severas restricciones sobre las
secuencias de aminoácidos posibles. Por ejemplo (Figura 19.43), al
aminoácido codificado por el triplete AUU sólo podría seguirle uno
codificado por un triplete UUX (siendo X cualquier base nitrogenada).
Estudios sobre la secuencia de aminoácidos de algunas proteínas
realizados por S. Brenner en 1957 pusieron de manifiesto que en la
naturaleza no existían tales restricciones, por lo que el esquema de
Gamow hubo de ser definitivamente descartado.
Un esquema alternativo para el código genético fue propuesto por F. Crick y sus colaboradores. Se trataba de un código sin solapamientos (cada triplete estaba implicado en la codificación de un solo aminoácido) y que proporcionaba una solución brillante al problema, no abordado por Gamow, de la necesidad e “símbolos espaciadores” entre los tripletes. Tal problema se ilustra en el siguiente ejemplo: Sea la secuencia ….UCGUGGGCAAUU…; podemos suponer que se traducirá de manera que los tripletes … UCG, UGG, GCA, AUU… codificarán unos determinados aminoácidos. Sin embargo, si la pauta de lectura de los tripletes se desplazase una sola base nitrogenada tendríamos … U, CGU, GGG, CAA, UU…., que evidentemente codificarían unos aminoácidos diferentes. ¿Cómo reconocen las células vivas cuál es la pauta de lectura “correcta”? ¿Existen símbolos espaciadores que delimitan los tripletes correctos evitando cualquier lectura alternativa? En el esquema de Crick este problema se solucionaba con un “diccionario de palabras con sentido”. Sólo un número limitado de tripletes del código codificarían aminoácidos; el resto serían tripletes “sin sentido” o no codificantes que tendrían el papel de evitar las lecturas incorrectas. En nuestro ejemplo, si UCG y UGG son codificantes, necesariamente CGU y GUG deberán ser “sin sentido”. Los cálculos de Crick demostraron que era perfectamente posible un código de estas características, en el que 20 de los tripletes codificarían a los 20 aminoácidos de las proteínas (no habría pues tripletes sinónimos) mientras que los 44 restantes serían “sin sentido”. El esquema de Crick explicaba además el sentido biológico del exceso de tripletes con respecto al de aminoácidos.
El código propuesto por Crick, sin duda de una gran elegancia, no es el que en realidad funciona en las células vivas. El curso de la evolución biológica parece haber optado por un esquema menos sofisticado y quizás más imperfecto. Las investigaciones que en los años 60 del siglo XX condujeron al descifrado del código revelaron también que el “diccionario de palabras sin sentido” no era necesario; los tripletes se leen simplemente de corrido, sin símbolos espaciadores, a partir de un triplete concreto que funciona como “símbolo de iniciación”. Hay además muchos tripletes sinónimos (que codifican el mismo aminoácido).
En efecto, el definitivo conocimiento de las características del código genético hubo de esperar a que se pudiese descifrar uno a uno el significado de cada uno de los 64 tripletes que lo integran. Esta investigación, que constituye una de las etapas más apasionantes de la biología molecular, se realizó en los primeros años 60 por tres grupos de investigación dirigidos por S. Ochoa, M. W. Nirenberg y H. G. Khorana (Figura 19.44).
En 1955 S. Ochoa y M. Grunberg-Manago habían descubierto un enzima, la polinucleótido fosforilasa, capaz de sintetizar RNA in vitro a partir de mezclas de ribonucleótidos difosfato sin necesidad de ningún tipo de molde según la reacción:
RNA (n) + NDP à RNA (n+1) + Pi
El enzima polimerizaba ribonucleótidos en secuencias al azar que dependían de la composición nucleotídica de la mezcla de reacción inicial. Es probable que la función biológica de este enzima esté relacionada más con la degradación del RNA a través de la reacción inversa que con su síntesis; sin embargo, la importancia de este descubrimiento residía en que la polinucleótido fosforilasa proporcionaba la posibilidad de obtener in vitro RNA mensajeros cuyas secuencias podían ser al menos parcialmente controladas.
Marshall Nirenberg consiguió en 1961 desarrollar sistemas libres de células que realizaban la síntesis de proteínas in vitro. En estos sistemas introdujo mRNA sintetizado por la polinucleótido fosforilasa y, una vez traducidos estos mensajeros artificiales, procedió a secuenciar los polipéptidos obtenidos. El primer ensayo consistió en añadir al sistema de traducción un mensajero constituido exclusivamente por nucleótidos de uracilo (poli-U). El resultado fue un polipéptido monótono del aminoácido fenilalanina. Evidentemente este aminoácido estaba codificado por el triplete UUU, que se convirtió así en la primera “palabra” del código en ser descifrada. Ensayos análogos permitieron descifrar el significado de los tripletes AAA (lisina), GGG (glicina) y CCC (prolina).
La siguiente etapa en el descifrado del código pasó por la obtención de mensajeros artificiales a partir de mezclas de dos ribonucleótidos. Por ejemplo, un mensajero sintetizado al azar a partir de nucleótidos de uracilo y citosina contendrá los tripletes UUU, UUC, UCC, UCU, CUU, CCU y CCC. El análisis de la secuencia de los polipéptidos obtenidos proporcionaba los primeros indicios de cuáles eran los aminoácidos codificados. A continuación, haciendo variar las proporciones entre los dos nucleótidos de la mezcla inicial y combinando la información procedente de ensayos con parejas de nucleótidos diferentes, se iba afinando el análisis hasta determinar qué triplete codificaba cada aminoácido. El procedimiento era extremadamente laborioso, y, además, la existencia de tripletes sinónimos daba lugar a algunas ambigüedades que el método empleado no permitía resolver. Sin embargo, la colaboración entre los equipos de Nirenberg y Ochoa permitió, en poco más de un año, conocer el significado de un buen número de tripletes.
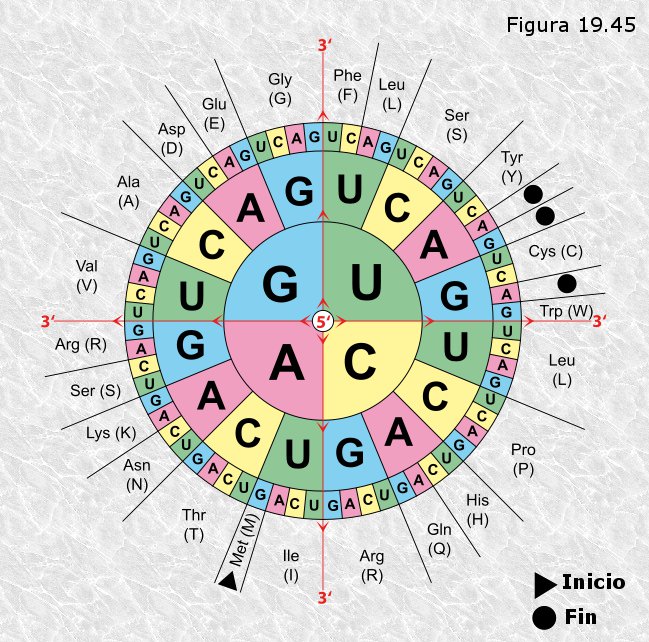 El descifrado de las últimas “palabras” del código resultó
especialmente complejo. Khorana consiguió fabricar mensajeros con dos
nucleótidos que se alternaban y más tarde con tripletes de secuencia
conocida que se repetían, lo que proporcionó bastantes claves
adicionales. Los esfuerzos culminaron con el descubrimiento por
Nirenberg de que trinucleótidos sintéticos de secuencia conocida podían
entrar en el ribosoma, haciendo el papel del mensajero, y fijar
selectivamente al tRNA correspondiente unido a su aminoácido
específico. Ello permitió descifrar los tripletes restantes y completar
el “diccionario” del código (Figura
19.45). Por convenio, el código genético se expresa en forma de
tripletes del mRNA leídos en dirección 5’à3’.
El descifrado de las últimas “palabras” del código resultó
especialmente complejo. Khorana consiguió fabricar mensajeros con dos
nucleótidos que se alternaban y más tarde con tripletes de secuencia
conocida que se repetían, lo que proporcionó bastantes claves
adicionales. Los esfuerzos culminaron con el descubrimiento por
Nirenberg de que trinucleótidos sintéticos de secuencia conocida podían
entrar en el ribosoma, haciendo el papel del mensajero, y fijar
selectivamente al tRNA correspondiente unido a su aminoácido
específico. Ello permitió descifrar los tripletes restantes y completar
el “diccionario” del código (Figura
19.45). Por convenio, el código genético se expresa en forma de
tripletes del mRNA leídos en dirección 5’à3’.
Las investigaciones que se han descrito fueron realizadas en la bacteria E. coli. Toda la experimentación posterior sustenta la idea de que el código genético es universal: los tripletes del código tienen el mismo significado en todas las células vivas tanto procariotas como eucariotas. Incluso se ha podido comprobar que sistemas de traducción in vitro obtenidos de una determinada especie son capaces de traducir mensajeros procedentes de especies diferentes. De todos modos, se han descrito algunas excepciones a la regla de universalidad del código, excepciones que afectan al significado de unos pocos tripletes en los genes de mitocondrias y cloroplastos.
Del conocimiento detallado del código se pudieron extraer como corolario algunas de sus características, lo que permitió corregir los modelos teóricos previamente elaborados por Crick:
El código genético:
-
No presenta solapamientos: cada triplete de bases está implicado en la codificación de un solo aminoácido.
-
Presenta tripletes sinónimos. Se suele aludir a esta propiedad diciendo que es un código degenerado.
-
Es un código “sin comas”: no presenta símbolos espaciadores. La lectura se realiza de corrido partiendo de un triplete de iniciación, que en la mayor parte de los casos es AUG. Este triplete codifica además el aminoácido metionina o alguno de sus derivados.
-
Es universal, aunque presenta un reducido número de excepciones, que afectan a la codificación de determinados tripletes en las mitocondrias.
5.5.- TRADUCCIÓN.
Como se ha comentado con anterioridad, el “dogma central” de la biología molecular afirma que la expresión génica es un proceso en dos etapas: transcripción y traducción. Analizado el primero de estos procesos y los pormenores del código que permite llevar a cabo el segundo, deberemos ahora analizar éste último detalladamente.
La primera cuestión que cabe plantearse acerca de la síntesis de las proteínas es el orden de ensamblaje de los aminoácidos. El procedimiento más sencillo para ensamblar un cierto número de aminoácidos consistiría en iniciar la cadena por uno de sus extremos e ir añadiendo aminoácidos secuencialmente hasta el extremo opuesto. Se ha podido comprobar, mediante experimentos con aminoácidos marcados radiactivamente, que la síntesis comienza por el extremo amino-terminal y finaliza por el extremo carboxi-terminal.
En segundo lugar cabe preguntarse acerca del tipo de interacción que se ha de establecer entre los ribosomas y el mRNA a la hora de llevar a cabo la síntesis. Dado que el ensamblaje de los aminoácidos es secuencial, no hay necesidad de que toda la cadena polinucleotídica del mensajero esté en contacto con el ribosoma en un momento dado, sino sólo la parte que codifica el aminoácido que se va a incorporar; es decir, el mensajero puede correr a través del ribosoma y ser leída su información de manera similar a una cinta magnetofónica que discurre por un cabezal de lectura. Por otra parte, no parece haber inconveniente para que el mensajero discurra simultáneamente a través de varios ribosomas que, concomitantemente, irían sintetizando varias cadenas polipeptídicas. Los primeros experimentos realizados acerca de la síntesis de proteínas confirmaron la corrección de estas ideas. Incluso se pudieron obtener, por medio de la microscopía electrónica, imágenes de grupos de ribosomas, los polirribosomas, unidos por una única molécula de mRNA que estaba siendo traducida por ellos.
Aclaradas estas cuestiones iniciales, quedaba por resolver el auténtico núcleo del problema, a saber, ¿qué mecanismo molecular es el responsable de que el ensamblaje de los aminoácidos se lleve a cabo en el orden dictado por los tripletes del mRNA?
Ya en 1958, antes por tanto de que se reconociese el papel del mRNA en el proceso, Francis Crick formuló algunas ideas acerca del mecanismo de la traducción que resultaron capitales para el desarrollo de la investigación posterior. En primer lugar, Crick rechazó, por considerarla poco plausible en términos físico-químicos, la idea de que la molécula patrón de RNA presentase unas cavidades en las que pudieran encajar las cadenas laterales de los distintos aminoácidos. Por el contrario, argumentó que la capacidad del RNA para servir como patrón debía residir en su capacidad para formar puentes de hidrógeno y que, por ello, debía existir una molécula adaptadora capaz de formar puentes de hidrógeno específicos con el RNA patrón, siendo esta molécula la encargada de llevar a los distintos aminoácidos a su posición correcta dentro de la cadena. Aún desconociendo la naturaleza de tal molécula adaptadora Crick pronosticó que debería contener nucleótidos e incluso aventuró que podría ser necesaria una serie de enzimas específicos que unirían cada aminoácido con su adaptador correspondiente. Una vez asumido que el código genético constaba de tripletes (o codones) de nucleótidos del mRNA, la hipótesis de Crick se complementó con la idea de que las moléculas adaptadoras debían presentar tripletes complementarios (anticodones) a través de los cuales interactuarían con el mRNA.
Las predicciones formuladas por Crick en su “hipótesis del adaptador” se cumplieron punto por punto. La búsqueda del adaptador en extractos de E. coli condujo al hallazgo del tRNA, cuyas características se han glosado con anterioridad en este capítulo. Pronto se constató que existían tantas especies moleculares de tRNA como tripletes con sentido integran el código genético y que una serie de enzimas, denominadas aminoacil-tRNA sintetasas, catalizan la unión específica de cada aminoácido con su molécula de tRNA. El papel de estos enzimas en el proceso de expresión génica es de especial importancia ya que constituyen el agente que “conoce” el código genético (el intérprete que domina el idioma de los nucleótidos y el de los aminoácidos). Hay que resaltar, sin embargo, que las propiedades de las aminoacil-tRNA sintetasas, como las de las demás enzimas, residen en su propia secuencia de aminoácidos, que en última instancia está también cifrada en el DNA, siendo este ácido nucleico el depositario último de la “esencia” del código.
En la actualidad se tiene un amplio conocimiento acerca del proceso de ensamblaje de los aminoácidos que integran una cadena polipeptídica, aunque en sus aspectos esenciales había sido ya elucidado alrededor de 1964. La secuencia de acontecimientos implicados es de una complejidad considerable y en ella participan agentes de naturaleza muy diversa, a saber, RNA mensajero, ribosomas, tRNAs, aminoácidos, moléculas energéticas, enzimas y una serie de factores de naturaleza proteica que intervienen en diferentes momentos. Distinguiremos en este proceso las siguientes fases: activación de los aminoácidos, iniciación de la cadena polipeptídica, elongación y terminación.
ACTIVACIÓN DE LOS AMINOÁCIDOS.
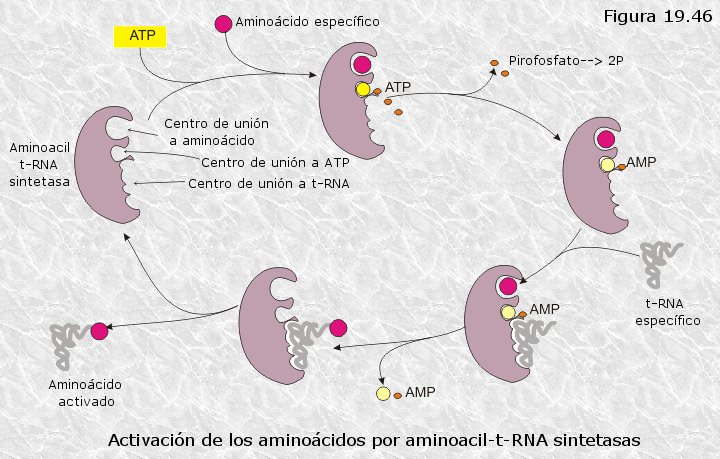
La activación de los aminoácidos consiste en su acoplamiento con su tRNA específico mediante una reacción catalizada por la correspondiente aminoacil-tRNA sintetasa. La unión se produce entre el grupo carboxilo del aminoácido y el grupo hidroxilo 3’ del nucleótido de adenina que se encuentra en el extremo 3’ del tRNA (Figura 19.46). La reacción requiere energía que es aportada por una molécula de ATP con liberación de un grupo pirofosfato.
INICIACIÓN DE LA CADENA POLIPEPTÍDICA.
La traducción de la secuencia de nucleótidos del mRNA a la correspondiente secuencia de aminoácidos comienza en el triplete de bases AUG, llamado codón de iniciación, que al mismo tiempo codifica el aminoácido metionina. En las células procariotas existen dos tRNA diferentes que se unen a este aminoácido, uno de ellos es el que interviene en la iniciación y en lugar de metionina transporta su derivado formil-metionina, el otro transporta metionina e interviene cuando este aminoácido se ha de incorporar en cualquier posición de la cadena polipetídica distinta de la inicial. Por el contrario, en las células eucariotas se incorpora metionina en ambos casos. Así pues, la síntesis de cualquier cadena polipeptídica empieza siempre por la metionina o por su derivado formilado, aunque en muchos casos estos aminoácidos son eliminados de la cadena en el llamado procesamiento post-traducción.

Puesto que es posible que en un mRNA haya más de un triplete AUG cabe preguntarse acerca del mecanismo que garantiza que la lectura comience en el triplete correcto. Se ha comprobado que en las células procariotas el mRNA presenta, entre el extremo 5’ y el primer triplete AUG, unas secuencias de bases que se aparean con otras complementarias de extremo 3’ de una molécula de rRNA que forma parte de la subunidad pequeña del ribosoma (Figura 19.47). Tales secuencias, denominadas “secuencias Shine-Dalgarno” aparecen conservadas en diferentes especies bacterianas y en bacteriófagos y su papel parece ser el de fijar al ribosoma en una posición próxima al triplete de iniciación antes de comenzar la lectura. En células eucariotas no se han detectado secuencias análogas y en ellas la traducción comienza simplemente en el triplete AUG más próximo al extremo 5’ del mensajero.
En la fase de iniciación se suceden los siguientes acontecimientos (Figura 19.48):
-
El mRNA se une a la subunidad pequeña del ribosoma con la intervención del factor IF3.
-
El tRNA iniciador previamente unido a su aminoácido específico (metionina o su derivado formilado) junto con el factor IF2 y una molécula de GTP se colocan de manera que el anticodón UAC del tRNA se aparea con el codón de iniciación del mensajero (AUG). El conjunto formado se denomina complejo de iniciación.
-
La subunidad mayor del ribosoma se une al complejo de iniciación. Se produce la hidrólisis del GTP a GDP + Pi y se liberan los factores de iniciación IF2 e IF3.
En el seno de la subunidad mayor del ribosoma se distinguen dos espacios destinados a acoger moléculas de tRNA: la sede P (peptidil) y la sede A (aminoacil). El tRNA de iniciación unido a formil-metionina queda situado en la sede P. Es posible que la formilación del grupo amino de la metionina tenga la misión de facilitar la entrada del tRNA de iniciación en la sede P “simulando” un enlace peptídico inexistente, pues en las sucesivas etapas de la traducción esta sede estará siempre ocupada por un tRNA unido a un péptido y no a un aminoácido.
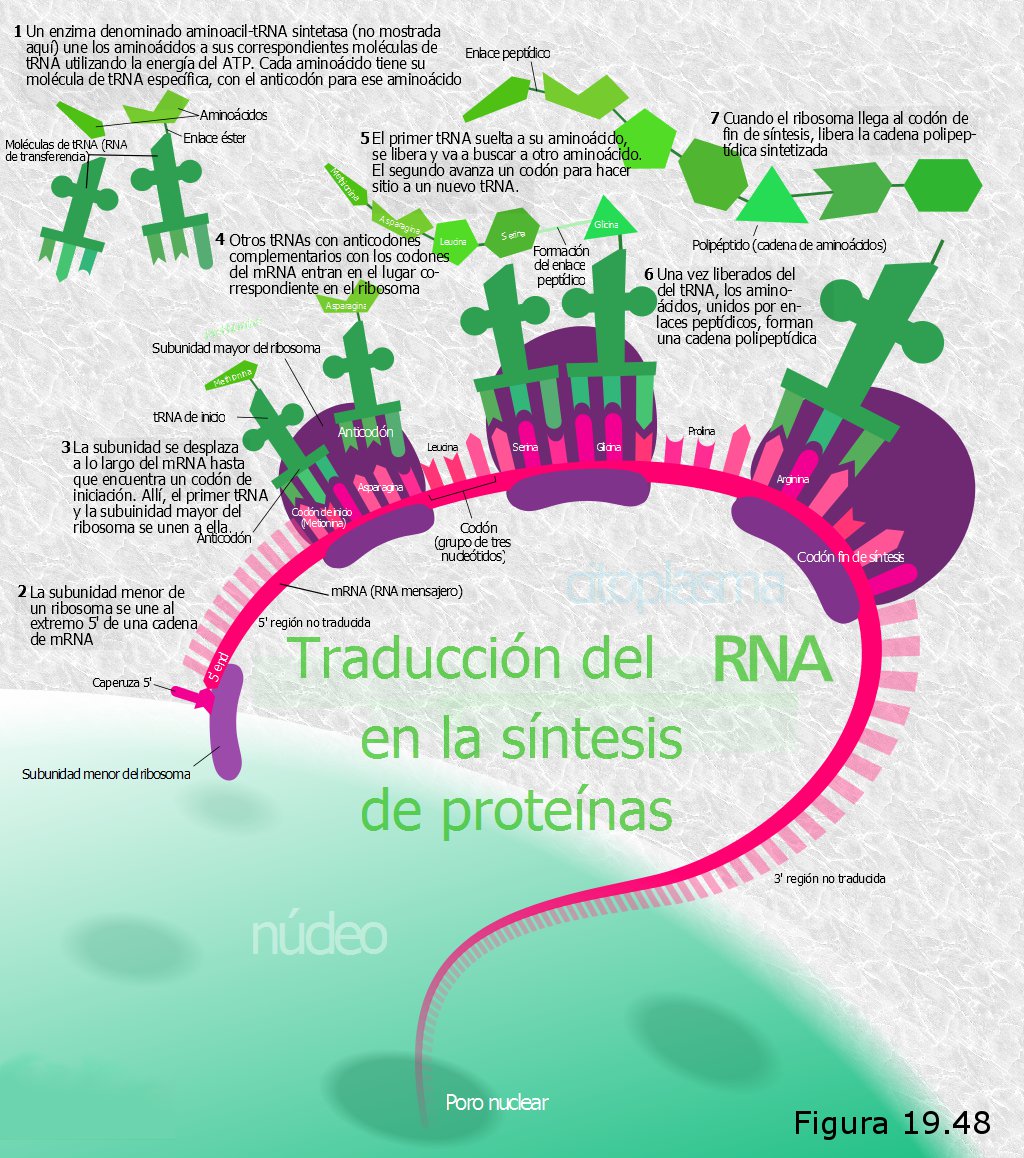
ELONGACIÓN DE LA CADENA POLIPEPTÍDICA.
En esta fase se precisa la concurrencia de varios factores proteicos de elongación (EF) y energía aportada por la hidrólisis del GTP. Se suceden los siguientes acontecimientos (Figura 19.48):
-
Un aminoácido activado entra en la sede A del ribosoma de manera que el anticodón del tRNA se aparea con el correspondiente codón del mensajero. Intervienen en el proceso el GTP y dos factores de elongación.
-
La formil-metionina unida al tRNA de iniciación se libera de éste y forma enlace peptídico a través de su grupo carboxilo libre con el grupo amino del aminoácido activado entrante que se encuentra en la sede A. La transferencia es catalizada por el enzima peptidil-transferasa. De este modo queda el tRNA de iniciación libre en la sede P y el tRNA del segundo aminoácido unido a un dipéptido en la sede A.
-
Se produce la traslocación del ribosoma, que se desplaza un triplete de bases del mensajero en dirección 5’à3’. Así, el tRNA unido a dipéptido queda situado en la sede P, quedando libre la sede A para la entrada de un nuevo aminoácido activado mientras que el tRNA de iniciación se libera del ribosoma. En el proceso interviene otro factor de elongación y se consume GTP.
-
Los pasos descritos se repiten tantas veces como aminoácidos haya que incorporar a la cadena polipeptídica, con la única diferencia de que en cada nuevo ciclo lo que transfiere la peptidil-transferasa en el segundo paso es ya un verdadero péptido (no formil-metionina como en la primera ocasión). El péptido transferido tendrá un aminoácido más de longitud en cada ciclo.
TERMINACIÓN DE LA CADENA POLIPEPTÍDICA.
La terminación de la cadena polipeptídica (Figura 19.48) se produce cuando el ribosoma llega a alguno de los tres codones sin sentido (UAA, UAG y UGA), que, por no corresponderse con ningún aminoácido, se interpretan como señal de “fin de síntesis”. Intervienen además varios factores proteicos de terminación (RF), los cuales, al no existir ningún tRNA que reconozca el codón de terminación, ocupan su lugar en la sede A, provocando la liberación por hidrólisis de la cadena polipeptídica finalizada, la disociación de las dos subunidades del ribosoma y la liberación del tRNA que quedaba en la sede P.
A la cadena polipeptídica así sintetizada sólo le resta adquirir su conformación tridimensional nativa para ser una proteína plenamente funcional e incorporarse a su destino en la maquinaria celular.
5.6.- REGULACIÓN DE LA EXPRESIÓN GÉNICA.
La alta eficacia que exhiben las células vivas en la manipulación de la materia y la energía se debe, entre otras cosas, a que funcionan atendiendo a un principio de economía molecular según el cual cualquier proceso que no sea necesario en un momento dado debe ser interrumpido. En un capítulo anterior ya hemos comprobado cómo las células llevan a la práctica este principio por medio de la regulación de su metabolismo a través de la actuación de distintos tipos de enzimas reguladores. Existe, sin embargo, un nivel superior de control de la actividad celular que es el de la síntesis de los propios enzimas implicados en el metabolismo y, en general, de todas las proteínas celulares. Resultaría un despilfarro, contrario al citado principio de economía, que una célula viva estuviese sintetizando continuamente todas las proteínas codificadas en su genoma con independencia de que estas fuesen necesarias o no en cada momento. La célula debe disponer de algún mecanismo que le permita reaccionar a las condiciones cambiantes del medio en que vive modificando la expresión de su información genética de la manera más adecuada.
Los primeros indicios de que las células controlan la síntesis de sus propios enzimas se tuvieron a comienzos del siglo XX cuando se comprobó que en las levaduras la presencia o ausencia de determinados azúcares en el medio de cultivo inducía la aparición o desaparición respectiva de los enzimas responsables de su metabolismo en el citoplasma celular. Pronto pudo comprobarse que los enzimas celulares podían dividirse en dos grandes clases: los enzimas constitutivos, presentes siempre en la célula pues son responsables de reacciones básicas que deben estar siempre en funcionamiento, y los enzimas adaptativos, que aparecen o desaparecen como respuesta a las condiciones cambiantes del medio.
Las investigaciones acerca de los mecanismos de regulación
de la expresión génica corrieron paralelas a las que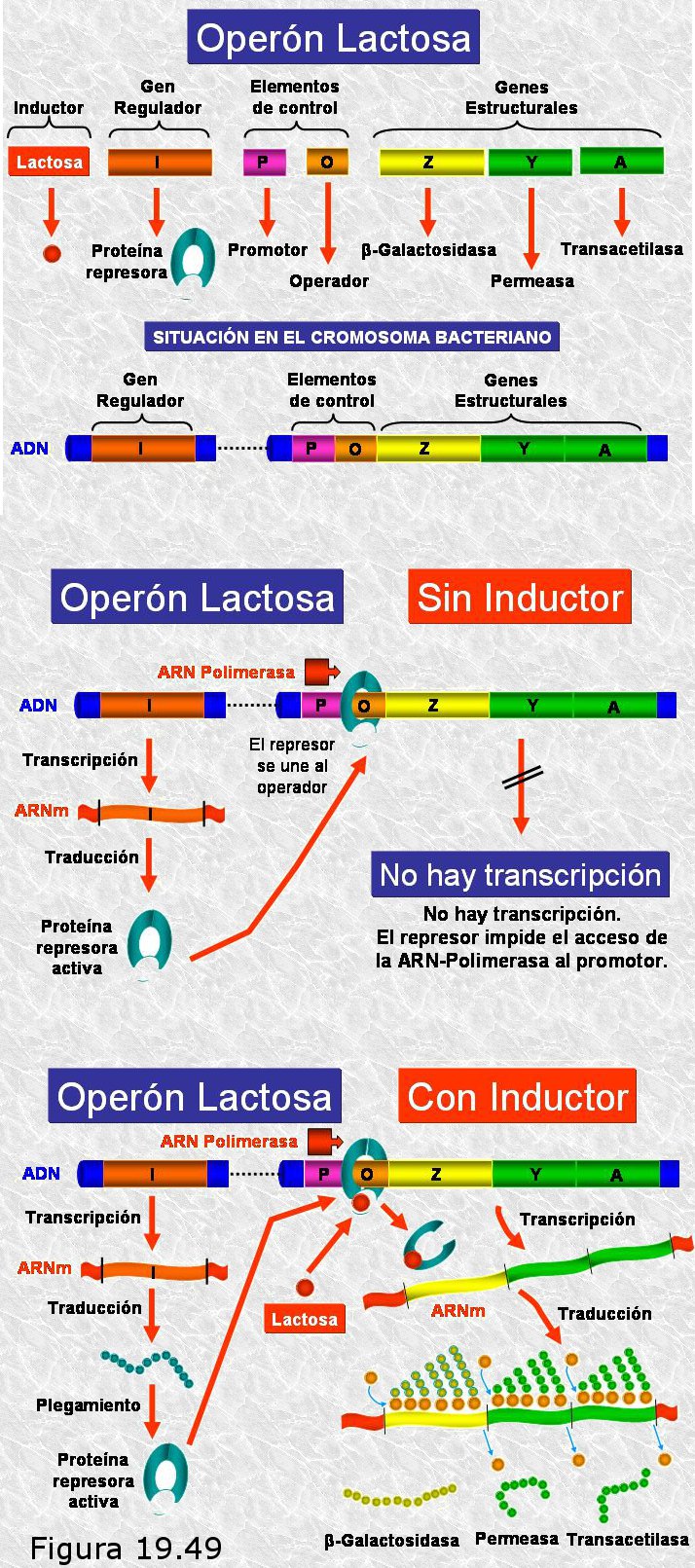 condujeron a la
comprensión del propio mecanismo de la expresión y se deben en gran
medida al trabajo de François Jacob y Jacques Monod (Figuras
19.39 y
19.38). Ambos científicos presentaron en 1961 un modelo, conocido
como el modelo del operón, que permitía comprender dichos
mecanismos reguladores. El modelo fue elaborado sobre la base del
análisis de un sistema enzimático inducible, presente en la bacteria
E. coli, del que forman parte los enzimas responsables de
metabolizar el azúcar lactosa.
condujeron a la
comprensión del propio mecanismo de la expresión y se deben en gran
medida al trabajo de François Jacob y Jacques Monod (Figuras
19.39 y
19.38). Ambos científicos presentaron en 1961 un modelo, conocido
como el modelo del operón, que permitía comprender dichos
mecanismos reguladores. El modelo fue elaborado sobre la base del
análisis de un sistema enzimático inducible, presente en la bacteria
E. coli, del que forman parte los enzimas responsables de
metabolizar el azúcar lactosa.
Un operón está constituido por los siguientes elementos:
-
Genes estructurales.- Son los genes sometidos a regulación. Codifican los polipéptidos funcionales que forman parte del sistema inducible. En general, los operones bacterianos incluyen varios genes estructurales (son policistrónicos)
-
Promotor.- Es una secuencia de DNA que es reconocida por la RNA polimerasa para iniciar la transcripción. Se encuentra adyacente a los genes estructurales, que comparten un único promotor.
-
Operador.- Es una secuencia de DNA situada entre el promotor y los genes estructurales. Se trata de un elemento regulador que determinará si se permite o no la transcripción de los genes estructurales.
-
Gen regulador.- Es un gen que codifica una proteína, denominada represor, con capacidad de permitir o bloquear la expresión de los genes estructurales. El gen regulador se encuentra cerca de los genes estructurales pero no necesariamente contiguo a ellos.
-
Represor.- Proteína codificada por el gen regulador. Interactúa de manera específica con el operador. Cuando se fija a éste bloquea la expresión de los genes estructurales impidiendo el avance de la RNA polimerasa. Cuando se disocia permite la transcripción del RNA mensajero correspondiente a los genes estructurales para su posterior traducción a polipéptidos.
-
Inductor/co-represor.- Metabolito que interactúa con el represor provocando, mediante cambios conformacionales, su disociación o su fijación al operador según los casos. Es la sustancia sobre la que actúa el sistema enzimático inducible o bien el producto final de la actividad de éste.
A continuación se describirán, a modo de ejemplo, los elementos que integran el operón lac, que fue el estudiado por Jacob y Monod, y los que integran el operón trp.
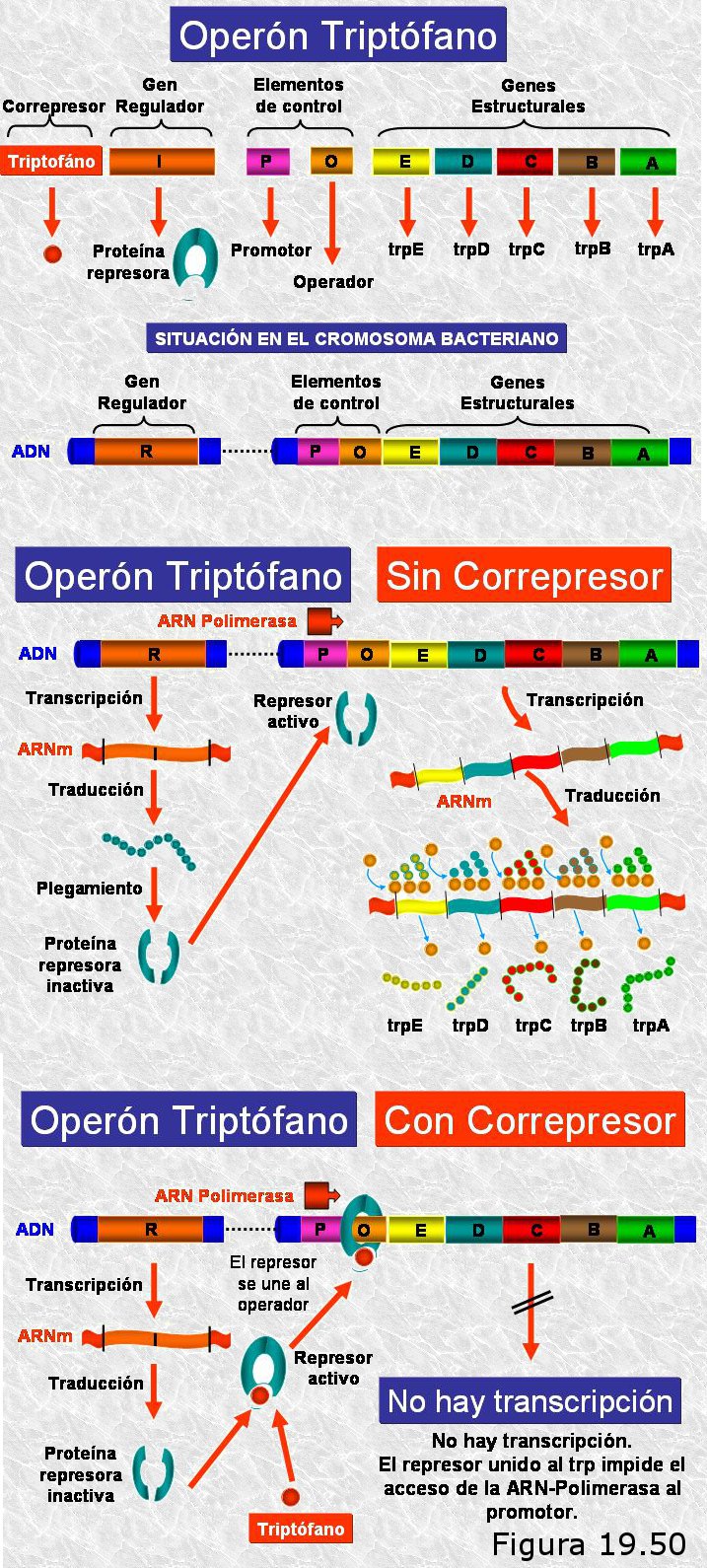 El operón lac (Figura
19.49) incluye tres genes estructurales: el gen Z, que
codifica en enzima β-galactosidasa, que cataliza la hidrólisis de
la lactosa a glucosa y galactosa; el gen Y, que codifica la
galactósido permeasa, una proteína de membrana que facilita el
acceso de la lactosa al interior celular; y el gen A, codifica la
galactósido transacetilasa, enzima cuyo papel en el metabolismo
de la lactosa no está del todo aclarado. El gen regulador i
codifica una proteína represora que, en ausencia de lactosa, se fija de
manera específica sobre el operador bloqueando la expresión de
los genes estructurales. Cuando la lactosa está presente funciona como
inductor, interactuando con el represor y provocando su
disociación del operador y desbloqueando así la expresión.
El operón lac (Figura
19.49) incluye tres genes estructurales: el gen Z, que
codifica en enzima β-galactosidasa, que cataliza la hidrólisis de
la lactosa a glucosa y galactosa; el gen Y, que codifica la
galactósido permeasa, una proteína de membrana que facilita el
acceso de la lactosa al interior celular; y el gen A, codifica la
galactósido transacetilasa, enzima cuyo papel en el metabolismo
de la lactosa no está del todo aclarado. El gen regulador i
codifica una proteína represora que, en ausencia de lactosa, se fija de
manera específica sobre el operador bloqueando la expresión de
los genes estructurales. Cuando la lactosa está presente funciona como
inductor, interactuando con el represor y provocando su
disociación del operador y desbloqueando así la expresión.
Un modelo alternativo al del operón lac es el del operón trp (Figura 19.50), integrado por un grupo de genes estructurales (genes E, D, C, B y A) que codifican los enzimas responsables de la síntesis intracelular del aminoácido triptófano. En este caso el represor libre resulta inactivo y no puede unirse al operador, quedando desbloqueada la expresión de los genes estructurales. Cuando el triptófano está presente en el medio intracelular actúa como co-represor, provocado en el represor un cambio conformacional que le permite unirse al operador y bloquear así la expresión de dichos genes.
En general, los sistemas inducibles, como el operón lac, son propios de las rutas catabólicas mientras que los represibles, como el operón trp, son propios de las rutas anabólicas.
Los sistemas de control hasta aquí descritos comparten la característica de que su acción se basa en una proteína represora que bloquea la expresión génica. Se han descrito también en células procariotas sistemas de control basados en la acción de proteínas activadoras que se denominan sistemas de control positivo. Las proteínas activadoras, también llamadas CAP (proteína activadora por catabolito) se unen al DNA en un lugar adyacente al promotor, el sitio CAP, de manera que favorecen la fijación de la RNA polimerasa facilitando así el inicio de la transcripción (Figura 19.51). Para que se pueda producir la unión al DNA de la proteína CAP ésta debe interactuar, a través de un centro alostérico, con el nucleótido cAMP (AMP cíclico), presente en el citoplasma celular. Los niveles intracelulares de cAMP están a su vez modulados, de manera todavía no bien conocida, por la presencia de determinados catabolitos.
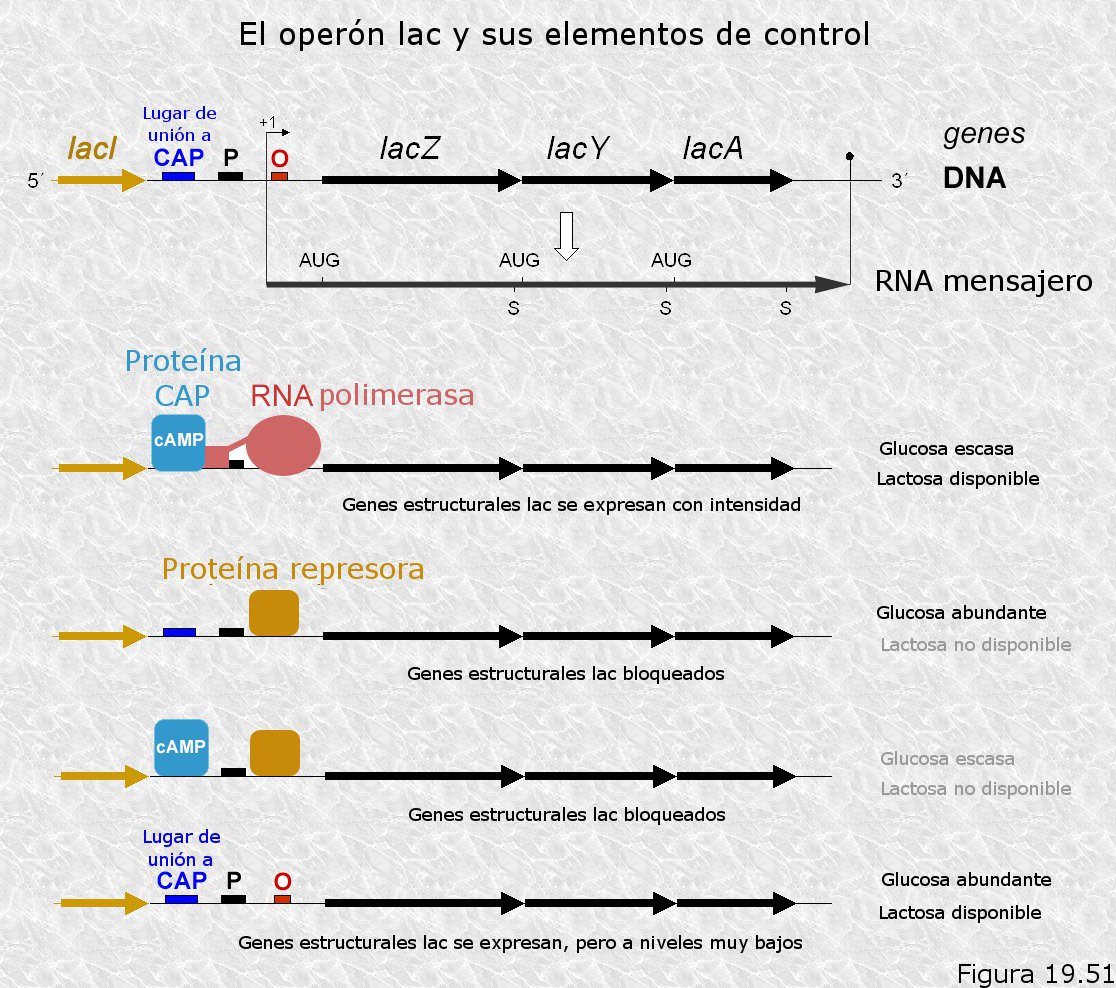
Un caso bien conocido de control positivo por CAP afecta a varios operones relacionados con el catabolismo de azúcares distintos de la glucosa, entre ellos el operón lac. Cuando E. coli crece en un medio rico en glucosa utiliza este azúcar como fuente de energía con preferencia frente a otros azúcares aunque también estén presentes en el medio de cultivo. Para ello la célula bloquea la expresión de los genes estructurales de los operones relacionados con estos azúcares. La presencia de glucosa hace descender los niveles de cAMP haciendo que CAP no se pueda fijar al DNA, con lo que la eficacia de la transcripción disminuye en todos los operones controlados, aun en el caso de que se encuentren desbloqueados por los respectivos represores. El mecanismo descrito se conoce con el nombre de represión por catabolito. La manera en que la presencia de glucosa afecta a los niveles intracelulares de cAMP no se conoce todavía con exactitud.
De lo dicho hasta ahora se deduce que la regulación de la expresión génica puede responder a alguno de los siguientes modelos teóricos:
-
Tipo 1.- Sistema inducible con control negativo (ej. Operón lac)
-
Tipo 2.- Sistema inducible con control positivo. (ej. Operones regulados por CAP)
-
Tipo 3.- Sistema represible con control negativo. (ej. Operón trp)
-
Tipo 4.- Sistema represible con control positivo. (No hay casos conocidos)
La regulación de la expresión génica en células procariotas es un fenómeno complejo en el que multitud de operones se ven sometidos a sistemas de control mixto en los que intervienen más de uno de los tipos reseñados. Por ejemplo el operón lac responde a los tipos 1 y 2.
Por otra parte, la regulación de la expresión génica e las células eucariotas y, más concretamente, en los eucariontes pluricelulares, es todavía, si cabe, más necesaria que en las células procariotas. En un organismo pluricelular todas las células poseen la misma información genética, pero esta información no se expresa por igual en todas ellas. La esencia de la pluricelularidad es la especialización: los distintos tipos celulares se van diferenciando a lo largo del desarrollo y adquiriendo cada uno capacidades y funciones diferentes. Esta especialización está basada en una expresión diferencial de la información genética, que se consigue mediante un sofisticado sistema de regulación.
Los mecanismos de regulación de la expresión génica en las células eucariotas se conocen con menor detalle que los de las células procariotas. Los mRNA eucariotas son monocistrónicos, lo que lleva a la conclusión de que los genes estructurales en estas células no están integrados en operones como en el caso de las procariotas. Por el contrario, cada gen eucariota dispone de su propio sistema regulador que no comparte con otros genes. Tales sistemas se componen de unas secuencias específicas denominadas enhancers, situadas en las proximidades del gen estructural, que pueden interactuar con varias proteínas de las llamadas factores reguladores de la transcripción. El número y tipo de los factores interactuantes, así como las secuencias enhancer implicadas determinan en cada caso la intensidad con la que se llevará a cabo la transcripción.
Sin duda es mucho lo que queda por investigar en el campo de la regulación de la expresión génica en organismos eucariontes y muchas las aplicaciones que puedan tener los conocimientos que sobre ello se vayan obteniendo.
6.- LA MUTACIÓN A NIVEL MOLECULAR.
Una de las implicaciones del reconocimiento del DNA como el material genético fue la comprensión del fenómeno de la mutación a nivel molecular. Asumido que la información genética se encontraba cifrada en forma de secuencia de nucleótidos se dedujo fácilmente que la mutación, al menos la mutación génica o puntual, consiste en una alteración de dicha secuencia de nucleótidos. Las mutaciones más simples son las que afectan a un único par de bases del DNA, que constituye la unidad mínima de mutación. En relación con el tipo de sustitución que afecta a un determinado par de bases se pueden distinguir dos tipos:
-
Transiciones.- Cambio de una base púrica por otra del mismo tipo o de una base pirimídica por otra del mismo tipo (AT—›GC, GC—›AT, CG—›TA y TA—›CG).
-
Transversiones.- Cambio de una base púrica por una pirimídica o viceversa (AT—›CG, AT—›TA, GC—›TA, GC—›CG, TA—›GC, TA—›AT, CG—›AT y CG—›GC).
Ambos tipos de sustitución se consolidan en el siguiente ciclo replicativo cuando la base sustituida forma parte del molde y enfrenta una base diferente de la original.
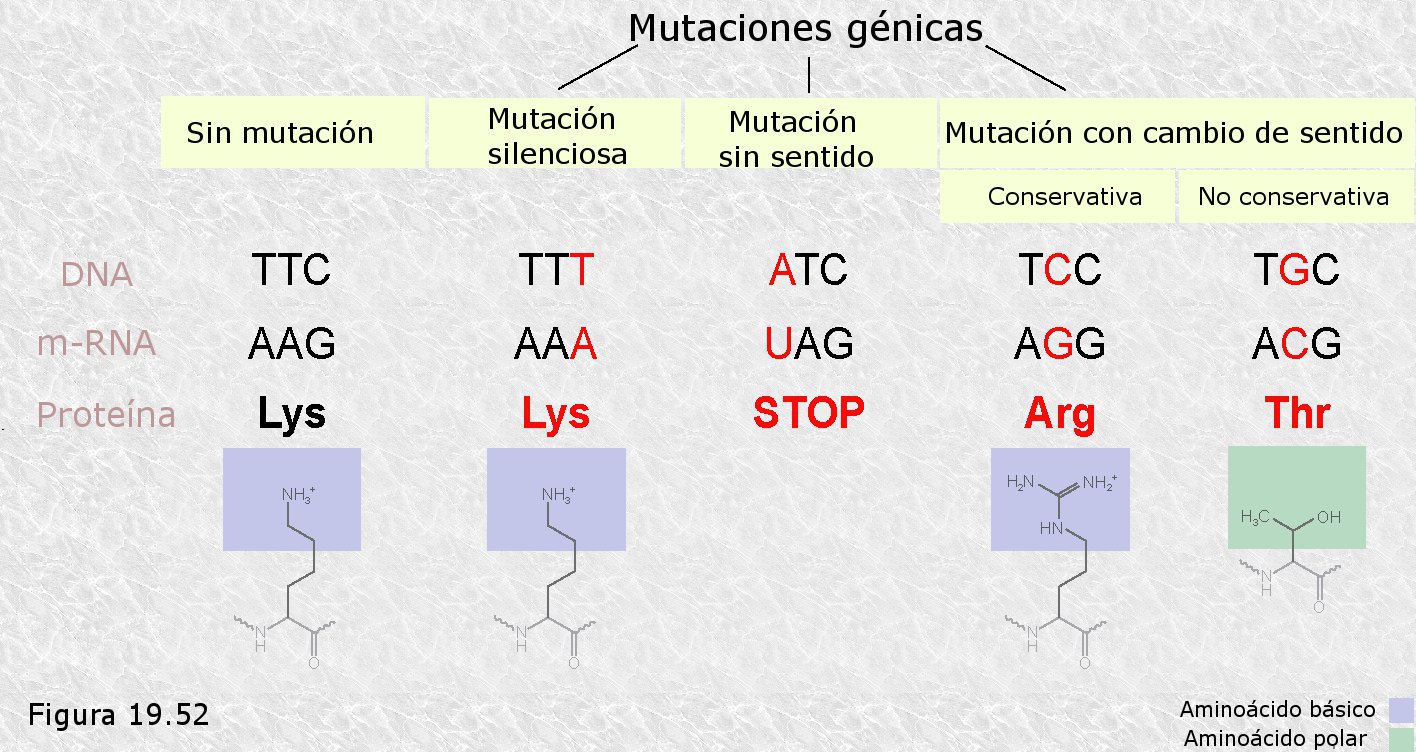
En cuanto a su efecto sobre el fenotipo se distinguen varios tipos de mutaciones, a saber:
Mutaciones con cambio de sentido.- Implican la sustitución de un aminoácido por otro. Su efecto sobre el fenotipo es moderado cuando ambos aminoácidos tienen propiedades químicas similares (mutación conservativa o neutra), y más drástico cuando sus propiedades difieren en mayor grado (mutación no conservativa).
Mutaciones sin sentido.- El cambio de base nitrogenada supone el cambio de un triplete que codifica un aminoácido por un triplete de fin de síntesis, lo que implica que a partir de este triplete no se insertan más aminoácidos en la cadena. El resultado, por lo general, es una proteína no funcional, por lo que este tipo de mutaciones suelen tener efectos severos sobre el fenotipo.
Mutaciones silenciosas.- No todas las mutaciones tienen un efecto sobre el fenotipo. Por el hecho de existir en el código genético tripletes sinónimos, algunas mutaciones no suponen un cambio en la secuencia de aminoácidos de la proteína codificada.
Por otra parte, algunas mutaciones puntuales pueden consistir no en sustituciones, sino en inserciones o pérdidas (deleciones) de pares de bases individuales. Estas mutaciones suelen tener efectos drásticos pues suponen un corrimiento de la pauta de lectura que, a la hora de sintetizar la proteína codificada, afecta a todos los aminoácidos que se encuentran después de la inserción o deleción.
En la Figura 19.53 se recogen algunas de las mutaciones génicas relacionadas con algunas enfermedades frecuentes en nuestra especie.
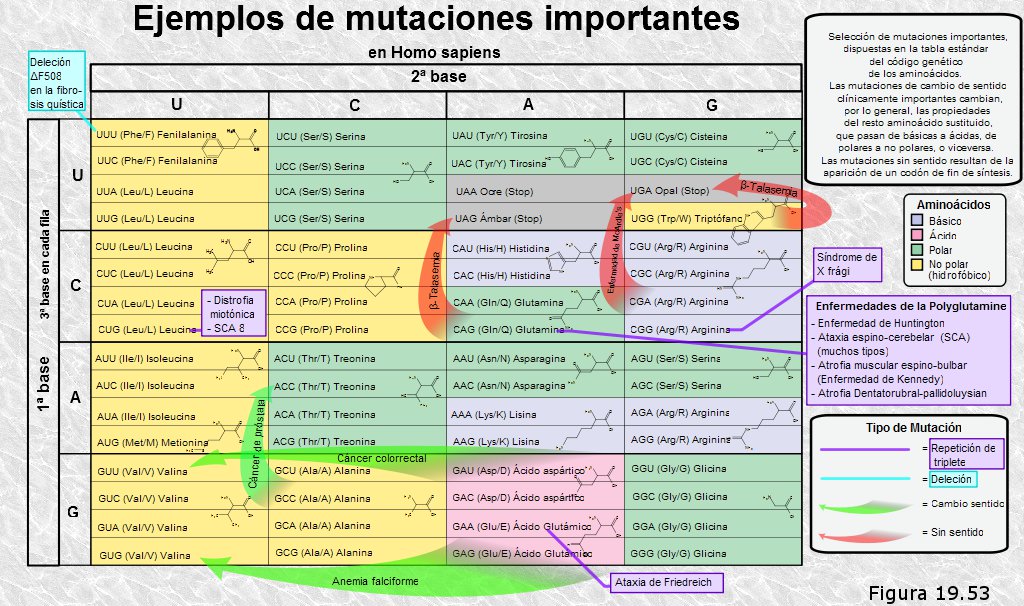
Podemos considerar, desde el punto de vista de sus causas, dos tipos de mutación: la mutación espontánea, que las células sufren en condiciones naturales, sin mediar la intervención de agentes externos; y la mutación inducida por agentes físicos o químicos externos a la célula.
6.1.- MUTACIONES ESPONTÁNEAS.
Las mutaciones espontáneas se producen fundamentalmente a causa de errores en la replicación del DNA, aunque también pueden obedecer a lesiones de carácter fortuito en moléculas de DNA que no se están replicando.
Los errores en la replicación pueden ocurrir mediante alguno de los dos siguientes mecanismos:
-
Tautomería.- Las bases nitrogenadas presentan un tipo particular de isomería denominado tautomería. La forma tautomérica más estable (forma ceto) coexiste en equilibrio con pequeñas cantidades de la forma menos estable (forma enol). Las formas inestables de las bases tienen alterada su capacidad para formar puentes de hidrógeno y dan lugar a apareamientos erróneos (A*-C, T*-G, G*-T y C*-A, donde el asterisco indica las formas inestables). Cuando el desplazamiento hacia la forma inestable se produce en el instante en que la base está siendo usada como molde en la replicación se produce un apareamiento erróneo que, si no es reparado, se consolida en el siguiente ciclo replicativo dando lugar a una transversión (Figura 19.54).
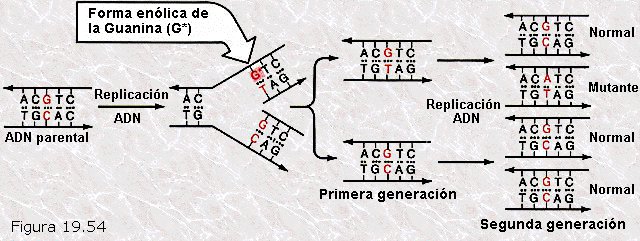
-
Apareamiento deslizado.- En secuencias muy repetitivas es relativamente probable que se produzcan un fenómeno denominado apareamiento erróneo deslizado, consistente en que alguna base se queda sin aparear formando un pequeño lazo. Si esto ocurre en la cadena en crecimiento se produce una adición de base mientras que si ocurre en la cadena molde se produce una deleción.
Por otra parte, las lesiones de carácter fortuito que puede sufrir el DNA suelen consistir en alteraciones químicas de las bases nitrogenadas que dan lugar a apareamientos erróneos los cuales se consolidan como mutaciones. Entre estas alteraciones cabe destacar la despurinización (pérdida de bases púricas que rompen su enlace N-glucosídico con la pentosa), la desaminación (pérdida de grupos amino afectando a la capacidad de formar puentes de hidrógeno) y daños oxidativos que resultan de la interacción de las bases con algunas sustancias como el peróxido de hidrógeno.
6.2.- MUTACIONES INDUCIDAS.
Existen diferentes tipos de agentes físicos y químicos con capacidad de producir mutaciones.
Entre los agentes físicos cabe destacar las radiaciones ionizantes como los rayos X o los rayos γ, algunas radiaciones no ionizantes como lo rayos ultravioleta y las emisiones radiactivas de partículas (α, β, neutrones). En general estos agentes actúan arrancando electrones a alguno de los átomos que forman parte del DNA, que al quedar ionizado se torna mucho más reactivo ante una amplia gama de sustancias presentes en la célula. Las reacciones químicas consiguientes pueden en alteraciones o pérdidas de bases nitrogenadas o incluso en la rotura de la molécula de DNA con pérdida de una parte del cromosoma.
Los agentes químicos con capacidad mutagénica pueden ser de varios tipos:
-
Análogos de base.- Sustancias con parecido estructural a las bases nitrogenadas que pueden sustituirlas en la replicación y dar lugar a apareamientos erróneos.
-
Modificadores de las bases.- Sustancias que afectan a las bases nitrogenadas alterándolas químicamente y modificando su capacidad para formar puentes de hidrógeno.
-
Agentes intercalantes.- Sustancias con estructura química planar o casi planar que se intercalan entre los pares de base durante la replicación dando lugar a adiciones o deleciones de base.
6.3.- REPARACIÓN DE LAS MUTACIONES.
Una parte significativa de las mutaciones que se producen en el DNA son reparadas mediante diferentes mecanismos de los que disponen las células. Durante el proceso replicativo la función correctora de las DNA polimerasas (exonucleasa 3’à5’) puede detectar y eliminar nucleótidos que hayan sido insertados de manera incorrecta. Algunas de las mutaciones no detectadas durante la replicación pueden ser detectadas más tarde y reparadas por un equipo de enzimas que incluye endonucleasas, exonucleasas, DNA polimerasas y ligasas.